



利用自洽场方法求解科恩-沈[吕九]方程,得到所设结构的晶体总能量(单点能)。在计算晶体的物理性质之前必须对所设晶体结构模型进行几何优化,根据关于能量、力、应力、位移的判据来判断晶体结构是否为稳定结构(总能量最小)。如果晶体结构不是稳定结构,重新设置晶格参数进行计算,直至得到稳定的晶体结构。对结构优化后的晶体进行物理性质计算,最后输出计算结果。
1.晶体的总能量
晶体总能量(不包括核的动能部分)可分成两部分:一部分是原子核与内层电子组成的离子实的能量,这部分能量基本上与晶体结构无关,是一个常数,赝势方法中常把总能量中这部分不变的能量设为零;另一部分是总能量与离子实能量之差,包括离子实与价电子的相互作用、离子实之间的相互作用以及价电子间的相互作用。晶体总能量与核动能(通常是零点振动能)之和与全部组分原子的孤立原子能之和的差,称为晶体的结合能。
在密度泛函理论中,晶体总能量ET是晶格电子的能量与离子实的排斥能之和
各项的物理意义与方程(8-11)和方程(8-12)中的相同。其中电子与外场v(r)的相互作用能为
电子间库仑相互作用能为
在局域密度近似条件下,电子的交换关联能为
离子实之间的库仑相互作用解为
这里Zs表示原子s的价电子数,R表示晶格平移矢量,τs表示原胞内原子s的相对位矢。动能泛函可通过科恩-沈[吕九]方程用单电子能量表示为
这里Ei是科恩-沈[吕九]方程的本征值。将式(8-50)~式(8-52)、式(8-54)和式(8-19)代入式(8-49),得到晶体总能量(https://www.xing528.com)
晶体的单电子能E与晶体体积NΩ之间的关系可用Murnaghan状态方程
来描述。方程(8-56)中N是晶体中原胞的数目,Ω是晶格常数尝试值为a时的原胞体积,B0是体弹性模量,B0′是B0对Ω的导数。对于不同的晶格常数尝试值a,即不同的晶体体积NΩ,可计算出相应的单点能E(NΩ),用最小二乘法拟合Murnaghan状态方程,便可得到相应于该结构的晶体常数a0、体弹性模量B0和该结构的能量极小值E(NΩ0)。
对于组成元素确定的体系,可能存在不同的晶体结构。分别对不同的晶体结构进行总能量计算和Murnaghan方程拟合,得到相应的能量极小值E(NΩ0),比较不同晶体结构的能量极小值,便可确定稳定的晶体结构。
由晶体总能量可以确定晶体的一些力学性质,如赫尔曼-费恩曼(Hellmann-Feynman)力和应力。作用于原子s上的力Fs可由晶体总能量ET对原子s的原胞内位矢τs求负梯度得到,即Fs=-∇τsET,这个作用于原子上的晶体内力称为赫尔曼-费恩曼力。对于晶格应变张量ε=εαβ,平均应力张量为σ=σαβ,则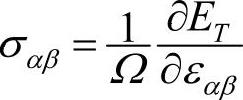 。
。
2.弹性常数
不同晶系的晶体具有不同数量的独立弹性常数。立方结构的晶体存在3个独立的弹性常数(C11,C12和C44),六方晶系具有5个独立的弹性常数(C11,C12,C13,C33和C44),而正交晶系则具有9个独立性常数(C11,C22,C33,C12,C13,C23,C44,C55和C66)。材料的弹性常数描述了它对所加应力的响应,或者反过来说,弹性常数描述了为维持一个给定的形变所需的应力。
为了计算晶体的弹性常数,可先对晶胞进行不同的弹性变形,然后算出变形后的能量,并通过此能量与未变形晶胞的能量差,求出弹性应变能,进而利用弹性常数与应变能的关系计算得到弹性常数。弹性应变能计算式为
其中ΔE为变形前后晶胞的能量差值;V0为原始晶胞的体积;Cij为弹性常数;ei和ej为应变。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。