



在实际的计算中,科恩-沈[吕九]方程是通过自洽计算来求解的,其计算流程如图8-4所示。图中的流程为:将多体系统原胞划分为足够细的网格点,在每个网格点上初始化一组试探波函数(通常设为随机数),然后算出网格上的科恩-沈[吕九]势,求解本征方程。解出来的本征函数的值与初始化的试探波函数的值一般不会相同,再将新解出来的波函数的一部分叠加到初始值上重新计算科恩-沈[吕九]势,利用修正过的势再次求解本征方程。所得到的本征函数又用于修正上一步循环输入的波函数……循环迭代的结果,是最终求得的本征函数的值不再变化,计算得以收敛。利用收敛后的这组单电子波函数,就可得到体系总能量和电荷密度分布。
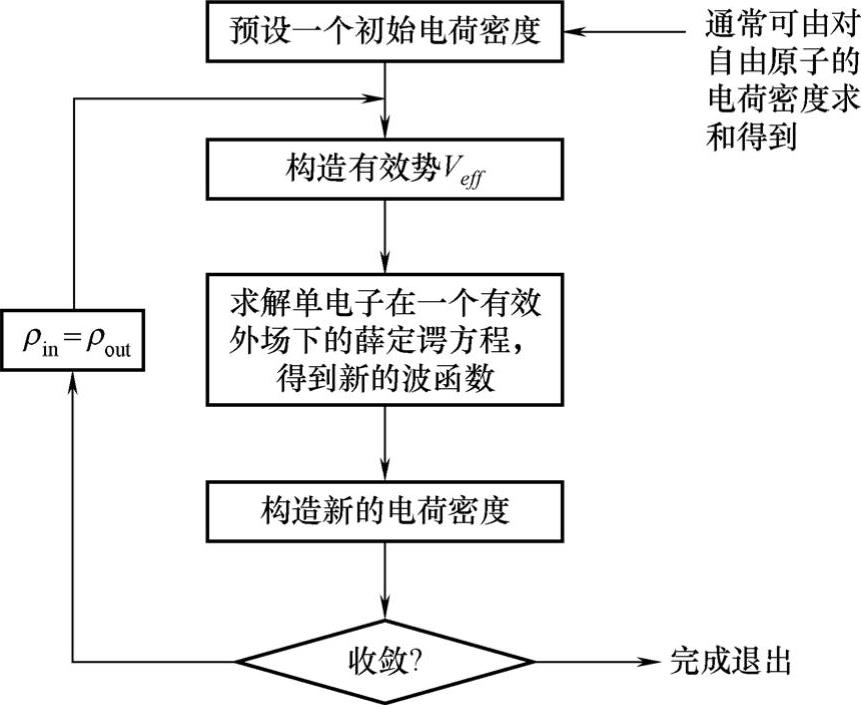
图8-4 自洽方法求解科恩-沈[吕九]方程流程图
1.求解单电子波函数
求解单电子波函数本质上是一个求本征值问题。对小矩阵,有很多数值算法可以直接求解。然而,电子结构计算中通常遇到的是很大的矩阵,无法直接对角化。当只对少数最低的本征值感兴趣时,可以用迭代算法求解。常见的迭代算法有,单带共轭梯度法、Davidson法与RMM-DIIS法以及Lanczos法,这里不再详细叙述,请参阅其他有关资料。
2.求电荷密度与能量
(1)分数占据(Partial Occupation)
在前面讨论布里渊区积分时,没有考虑占据数(零温下为阶梯函数)的影响。
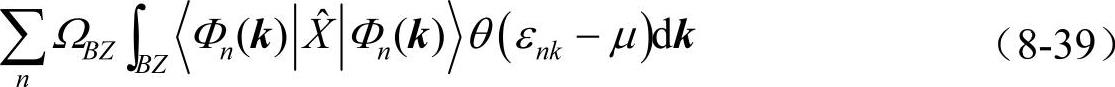
事实上,阶梯函数在数值上不稳定,使得布里渊区积分随k点数目收敛很慢。此时,常引入分数占据来代替阶梯函数。
1)费米-狄拉克Fermi-Dirac分布函数

优点:温度具有物理意义。
2)高斯(Gauss)型

优点:可以外推得到T=0时的能量。
3)Methfessel&Paxton方法。MP方法通过将阶梯函数用一组正交函数展开来实现,Gaussian函数是其零阶近似。因此,我们通常说的MP方法是指大于0阶的情形。对于合理的σ=kBT的值,MP方法中F里的熵贡献较小,这样计算的力会更准。一个合理的判据是选择合适的σ,使得熵小于1meV/(atom·K)。MP方法的弱点是不适用于半导体或绝缘体,有可能出现占据数大于1的情况。
4)四面体(Tetrahedron)方法
数占据从算法上也可以看成对布里渊区积分离散化时,对不同k点的权重进行系统的调整。从这个思路出发,在由四个k点组成四面体内做线性插值可以得到修正的权重,也即等效的分数占据函数fnk。实际应用上常采用Blochl等人提出的修正的四面体算法。
1)自动选择四面体使误差尽量抵消。(https://www.xing528.com)
2)对金属,误差抵消不完全,加入修正项。
3)最后积分形式仍为加权求和。
修正的四面体算法可以十分准确地计算能量、态密度等量,且不需要输入额外的参数。但是,由于它对分数占据不满足变分原理,所以得到的力可能不够准确,不能用来优化金属体系。
(2)能量泛函
在SCF迭代过程中,运用类似与热力学中的勒让德(Legendre)变换,能量泛函可以写成不同的形式。收敛以后它们都得到同一个能量,但是它们趋向于收敛的行为各不相同。KS能量泛函的原始定义为
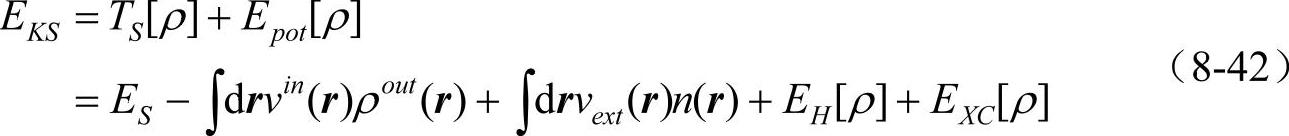
KS能量泛函原则上是电荷密度的泛函,但是在实际操作中可以看成有效势的泛函
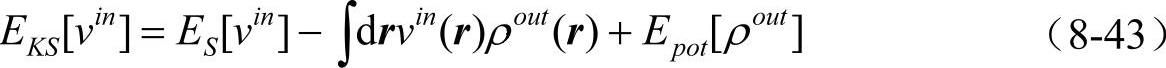
它满足变分原理。我们也选择另外一种显式的密度泛函形式,这就是常用的Harris-Foulkes(HF)泛函。因为输入势可以由输入密度决定,我们将v[ρin]记为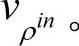
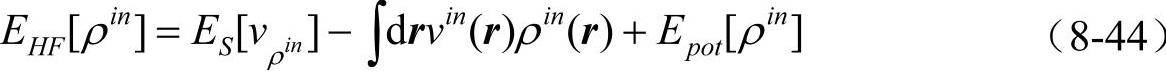
因为自洽收敛时ρin=ρout,HF泛函显然与KS泛函有同样的收敛性质。但是,HF泛函不满足变分原理,它主要用在非自洽计算(无需计算ρout)时的能量估计,而且在自洽计算的过程中HF泛函给出的能量值也通常比KS泛函更接近真实能量。
3.SCF迭代:mixing
SCF迭代刚开始时ρin和ρout可能会相差很大,使得整个算法不稳定,收敛困难。这时需要做一定的mixing。
(1)Linear mixing

为了考察的α取法,定义与真实密度的差为δρ=ρ-ρKS。在基态密度附近线性化,δρout=(χ~+1)δρin,若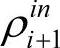 =ρKS,则可收敛。可以证明,欲使SCF收敛,则有:
=ρKS,则可收敛。可以证明,欲使SCF收敛,则有: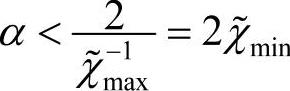 。
。
从物理上来看,响应函数可以看成极化率的度量。所以,对束缚很强的刚性系统,可以取较大的α,而对于金属表面之类的较软的系统,则比较困难。
(2)其他Mixing算法
对介电函数作均匀电子气Thomas-Fermi屏蔽近似,可以得到Kerker Mixing,其形式与linear mixing基本相似,这里不作具体介绍。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。