



1.原子间的作用力
两个原子间的化学亲和力,从物理本质上说,是由于在这两个原子之间存在吸引力。然而即使在化学亲和力很强的情况下,原子也难以过于接近,因为当原子相距很近时,它们之间主要显示排斥力。这种排斥力是一种短程力,即在远距离处它的数值很小,但在近距离处它比吸引力大。在平衡状态时,原子间的吸引力和排斥力相等。这时两个原子间的距离即为平衡距离。物体所受的合力为势能随距离的改变率。因此,原子在平衡距离处的势能具有极小值。
一个材料内部的势能等于每个原子的能量及相互作用能量的和。势能的零点可以任选,令原子相距很远,都处于基态,相互没有作用时,其势能U=0。那么要形成一个固体,必须把这些原子从远处移到一起,假如所形成的原子集团,要比原来分散的原子更稳定,则其势能必须小于零,就是说总的相互作用能必须是负值。由于原子在吸引时做功,所以吸引的势能Ua是负值,它是原子间距离r的幂函数。Ua=-α/rn,α为正的比例常数。当距离很远时,Ua=0,以后随距离缩短而Ua绝对值减小,如图2-8a最下面的虚线所示。相反,排斥的势能Ur是正值,要把互相排斥的原子聚在一起,外界必须对它做功。Ur的大小也是原子间距离r的幂函数幂。Ur=β/rm,如图2-8a上面虚线所示,β也为正的比例常数。当r减小到一定程度时,Ur急剧增加,致使Ur比Ua变化得更快。总的势能U是该两项值的和,即
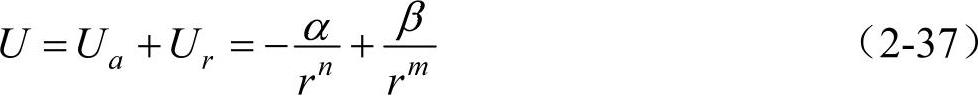
U在图2-8a中以实线表示。
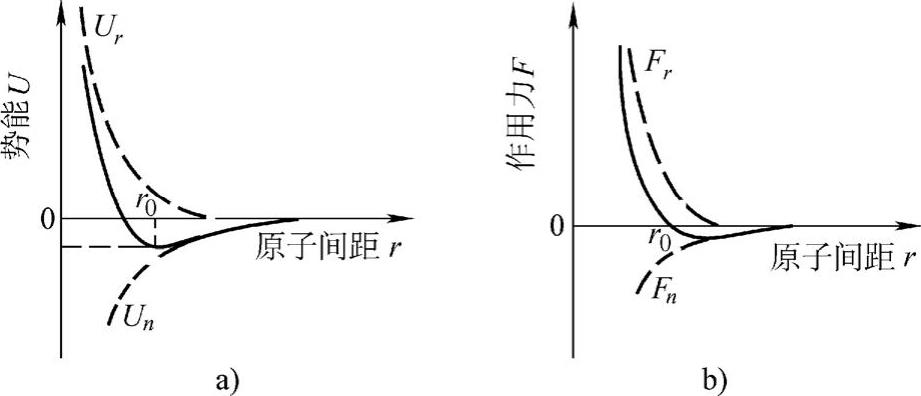
图2-8 势能及作用力与原子间距离关系
a)势能曲线 b)作用力曲线
原子间的作用力F(r)可由式(2-37)微分导出
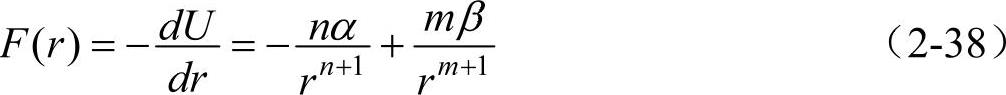
式中,m>n,负项为引力Fa;正项为斥力Fr,见图2-8b。当达到平衡间距r0时,吸引力和排斥力平衡,即F(r0)=0,则

r=r0时,原子集团的势能具有最低值,即在图2-8a中如果增加或减小原子间距离,都使势能增加,原子集团变得不稳定。对于某些离子晶体,n=1,m=9;对于某些金属,n=1,m=3;对于其他类型的晶体,n和m又有不同的数值。
可以设想固体的一个分子是这样的:在它中间有两个以上的原子核,外面绕有负电荷的电子云,靠静电引力结合起来。其结合强度取决于原子核之间各点的电子云密度。(https://www.xing528.com)
2.原子间作用力的由来当原子由远处移近时,在原子之间产生引力。在原子间距达到几个 (
(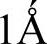 =0.1nm)时,产生了平衡引力的斥力。斥力的产生最主要是由于原子核外面的电子云有了重叠。当两个电子壳层开始重叠时,每一壳层中的电子都不能再认为是仅属于其原来的原子了,而为分子整体所共有。在这些壳层中的电子的运动和能量仍然遵守量子规律,特别是不相容原理。如果这两个电子壳层原来都是填满的,那么当它们结合成一个统一的电子集团时,不能保证所有的电子都占据低能量的量子状态,其中有些电子就要攀登到较高的能级。这种能量的增加就引起了斥力。满壳层重叠时必然导致上述的斥力。在价电子的电子云没有重叠以前,中性原子间不会有很强的化学结合力。
=0.1nm)时,产生了平衡引力的斥力。斥力的产生最主要是由于原子核外面的电子云有了重叠。当两个电子壳层开始重叠时,每一壳层中的电子都不能再认为是仅属于其原来的原子了,而为分子整体所共有。在这些壳层中的电子的运动和能量仍然遵守量子规律,特别是不相容原理。如果这两个电子壳层原来都是填满的,那么当它们结合成一个统一的电子集团时,不能保证所有的电子都占据低能量的量子状态,其中有些电子就要攀登到较高的能级。这种能量的增加就引起了斥力。满壳层重叠时必然导致上述的斥力。在价电子的电子云没有重叠以前,中性原子间不会有很强的化学结合力。
当两个原子相互移近时,距原子核最远的价电子比其他电子更早发生重叠,因为价电子只占据部分填充的壳层,当它们重叠时就会产生引力。但是,当两个原子满壳层的电子云也发生重叠时,就会出现满壳层间的斥力。
价电子重叠所产生的引力,可以用氢分子的形成来说明。氢分子由两个质子和两个电子所组成。在分析电子的运动时,质子可以认为是静止的,其作用力主要考虑粒子之间最强的静电力。如果这些粒子之间的力要形成化学键,则两个电子必须把大部分时间用在两个核之间。当两个电子互相接近时,它们之间的斥力是大的;但同时,由于这两个电子同时接近两个质子,因而产生四个引力,这四个引力足够抵消上述斥力。用量子力学处理这个问题,可以知道电子在分子的各个地方运动所用去的时间,从而计算出这四个粒子之间静电力的平均值。如果电子把大部分时间用在分子中的某个地方,那么,在计算平均作用能量时,这个地方的电子与其他粒子作用的能量就较大。
电子在分子中的分布是遵守量子力学规律的。假定最初原子相距很远,相互间没有作用,这时每个原子有一个电子处于(1s)状态。这个状态的波函数是一个以原子核为中心的球形分布的电子云。电子云密度随其与原子核的距离的增加而急剧减小。如把两个原子彼此移近,使它们的电子云恰好有不大的重叠,那么这时电子运动的特点是,电子能够从一个原子跑到另一个原子上去,而且不可能区分哪个电子是属于哪个原子。这样就改变了原来的波函数,原来是两个(1s)函数,现在需要用新的波函数来表示电子在分子整体中的运动。假设两个(1s)电子云开始重叠时没有什么变化,那么就有两个不同的(1s)型的ψ函数,以原子核a为中心有ψa,以原子核b为中心有ψb。两个电子用1和2表示,并且以ψa1表示电子1处于ψa的量子状态。这样,在分子中某点P的ψa12表示当电子1处于原子核a的1s状态时,它在P点出现的几率。同样,ψb22表示当电子2处于原子核b的(1s)状态时,它在某点Q出现的几率。因此,当电子2在Q点时,在P点找到电子1的几率为它们两者几率的乘积ψa12ψb22,这称为两者的合成几率。这就有可能把ψa1ψb2看作是两个电子的总的波函数。也可设想电子2在P点有波函数ψa2,电子1在Q点有波函数ψb1,那么就得到总的波函数为ψa2ψb1。显然,以上得到的两个波函数没有一个能单独满足要求,因为每个电子都能从一个原子移往另一个原子,并不能区分那个电子是属于那个原子的。我们所能知道的,只是当一个电子在Q点时,另一个电子在P点出现的几率,因此ψa1ψb2与ψa2ψb1必须有相同的机会出现。波函数的性质之一是可以把已知的波函数相加得出新的波函数。对于薛定谔方程来说,已知解的线性组合,仍为其解。因此得出新的波函数ψ+和ψ-为

在ψ+中把标号1和2互换,不改变波函数;在ψ-中交换1和2的标号,则ψ-为负值。所以ψ+是一个对称的波函数,而ψ-是一个反对称的波函数。
相应于式(2-40)和式(2-41)波函数的合成几率为
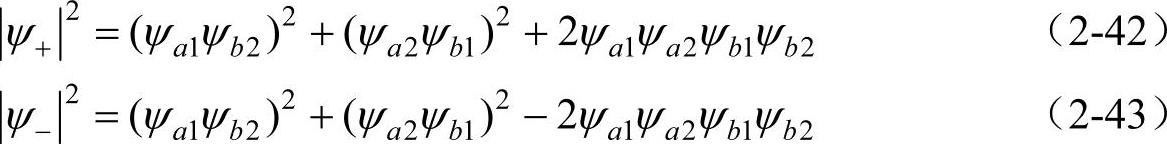
在式(2-42)和式(2-43)中,前两项的四个ψ函数是描述一个电子围绕一个原子核处于(1s)状态运动的波函数,它们的数值在两个原子核的中心处大致是相等的。两个方程的最后一项代表两个原子的交换作用,这是因为它们有交换各自电子的可能性。交换作用对分子能量的贡献通常称为交换能。所以在两个原子核的中心处│ψ+│2的值很大,而│ψ-│2等于零。因此ψ+所表示的是电子在分子中以大部分时间用在原子核之间,并且把它们联结在一起,形成一个稳定分子的量子状态;而ψ-则表示电子避开这个中心区域,不能形成结合键的一个状态。原来的(1s)能级就分裂成如图2-9所示的两个能级εA(r)及εs(r)。其中εs(r)这个能级属于ψ+,εs(r)曲线在r=0.87Å(0.087nm)处有一个能量最低点,说明两个氢原子接近到这种程度时可以形成稳定的H2分子,这就是结合状态(图中虚线为实验测定值),这时它们的两个电子是自旋相反的。当两个氢原子相互接近时,如果它们的电子自旋是平行的,那么原子间的相互作用永远是排斥的,如图2-9中εA(r)曲线,不能形成稳定的分子。εA(r)这个能级属于ψ-。
上面用氢原子价电子重叠形成氢分子解释了原子作用力的由来。这个原理同样可以用来说明金属理论中的一些问题。例如两个氢原子彼此接近时,1s能级分裂为两个能级的情况与金属价电子能级展宽是很相似的。氢分子只有两个氢原子,1s只能分裂成两个能级;而在金属晶体中,每立方厘米体积中具有1023数量级个原子,因此可分裂成间隔很小数量很多的能级,称为能带,但它们的本质过程是一样的。
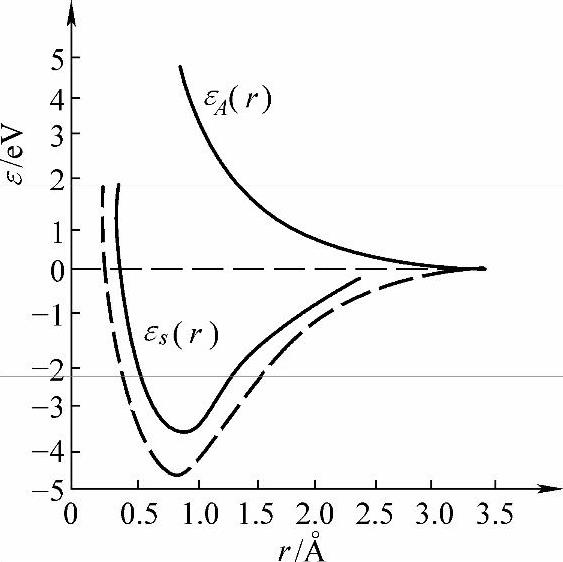
图2-9 氢分子1s能级分裂
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。