




图1-4 表面分子受力分析
1.液体的表面张力
分子在体相内部与界面上所处的环境是不同的。假设有一个液体表面分子,下边是本体相分子,图1-4所示为表面分子受力分析,在本体相中由于它与相邻单元的相互作用,该单元受到了一个均匀的力场。而在表面上单元受到的力不再是均匀的,尽管它将继续受到与其相互连接的本体相中相邻单元的吸引作用,但由于在表面,所以d方向的吸引较小。从而这个分子受到一个垂直于液体表面、指向本体内部的“合吸力”,通常称为净吸力。
由于有净吸力存在,致使液体表面的分子有被拉入本体内部减小表面的倾向,所以任何液体表面都有自发缩小的倾向,产生表面收缩力。表面分子在水平方向受到表面其他分子的吸引力,所以净吸力的结果是沿表面产生了横向张紧的状态。如果将作用在两个分子之间的力想象成弹簧,就能够把这种情况看成是这样的:在本体相中,分子的各个方向通过相等强度振动的弹簧被推拉,对一个特定的分子来说,时间平均的结果是一些平衡位置。在界面上,分子受到的拉入到本体相中弹簧力一般比邻相中弹簧的拉力强,结果界面分子被拉进本体,使表面区域分子的净密度减小。表面分子之间有较多的空间,在它们之间作用的弹簧的伸展远远超过了它们的平衡长度,因此产生一个沿着表面的张力使分子保持在一起。这种沿着表面的弹簧拉力,就是表面张力。从宏观层面来看,液体表面仿佛存在一层紧绷的液膜,在膜内处处存在的使膜紧绷的力就是表面张力。
观测表面张力最典型的试验是皂膜试验,如图1-5所示。图中所示为一带有活动金属丝的金属丝框ABCD。CD为可动边,边长为l。刚从皂液中提起这个金属框时,可观察到CD会自动收缩。要维持CD边不动,则需要施加适当的外力F。可见,CD边受到一个与力F大小相等、方向相反的作用,这个力与长度有关。皂膜有一定厚度(与分子大小相较不可忽略),表面的长度为2l。因此F与l就有如下关系
F=σ×2l (1-1)
式中 σ——比例系数,称为表面张力系数或比表面张力,一般简称为表面张力。
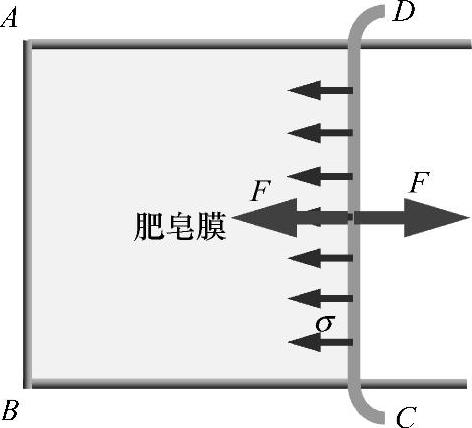
图1-5 皂膜试验
表面张力的方向和表面平行,表面张力也可认为是作用在金属丝框单位长度上的力。表面张力是体系的一种强度性质(此种性质不具有加和性,其数值取决于体系自身的特性,与体系的数量无关),受到多种因素的影响。σ除与液体性质及液面外相邻物质的性质有关外,还与温度及液体所含的杂质有关。能显著减小σ的物质称为表面活性剂。
(1)与物质本性有关 分子间作用力越大,σ越大,即σ金属键>σ离子键>σ极性键>σ非极性键。表面张力起源于净吸力,而净吸力取决于分子间引力和分子结构。因此不同的物质具有不同的表面张力,非极性有机液体的σ值一般较小,如正己烷在20℃下,σ=18.4 mN·m-1,因为其分子间相互作用力主要是色散力。对于极性分子,分子间的吸引力较大,表面张力较大,20℃常压下,水的表面张力为σ=72.8 mN·m-1。有金属键作用的液体,表面张力更大,汞在20℃下,σ=486.5 mN·m-1。现在已知表面张力最低的液体是氦,在1 K下σ=0.365 mN·m-1。表面张力最高的液体是熔融的铁,在其熔点1550℃下,σ=1880 mN·m-1。
(2)与接触相的性质有关 通常所说的某种液体的表面张力,是指该液体与含本身蒸气的空气相接触时的测定值。在与液体相接触的另一相物质的性质改变时,表面张力会发生变化。
在液-气界面上,表面张力源于液体分子相互吸引所产生的净吸力,空气分子对液体分子的吸引可以忽略。但在液1-液2界面上(相当于把空气换成了另一种液体),两种不同的分子也要相互吸引,因而降低了每种液体本体分子对表面分子的净吸力,使新界面的张力比原有的与空气接触时测得的小。
两个液相之间的界面张力是两液体已相互饱和(尽管互溶度可能很小)时两个液体的表面张力之差,即σ1,2=σ1′-σ2′。
(3)与温度有关 一般来说,温度升高,σ减小。非缔合性液体的σ-T关系基本上是线性的,即
σT=σ0[1-K(T-T0)] (1-2)
式中 σT、σ0——温度为T和T0时的表面张力;
K——表面张力的温度系数。
温度升高到临界温度Tc时,σ→0。温度升高时一般液体的表面张力都降低。当温度升高到接近临界温度Tc时,液-气界面逐渐消失,表面张力趋近于零。因为温度升高时物质膨胀,分子距离增大,故吸引力减弱,所以σ降低。
(4)与压力有关 一般来说,压力升高,σ减小。从气-液两相密度差和净吸力考虑,气相压力对表面张力是有影响的。因为在一定温度下液体的蒸气压是定值,因此只能靠改变其相中空气的压力或加惰性气体等方法来改变气相的压力。因为压力提高,气相密度增大,表面分子受力不均匀性略有好转。另外,若气相中有其他物质,则压力提高,促使表面吸附增加,气体溶解度增加,也使表面张力下降。所以通过改变空气或惰性气体压力所测得的表面张力变化应包括溶解、吸附、压力等因素的综合影响。综上所述,表面张力一般随压力的提高而下降,但影响不大。
2.液体的表面自由能
对于皂膜试验,从热力学角度来看,液膜在外力F的作用下移动了dx距离,做功δW=Fdx,结果是使表面积增加了dA=2ldx,如图1-6所示。
 (https://www.xing528.com)
(https://www.xing528.com)
图1-6 皂膜试验——表面自由能计算
恒T、p,恒组成,可逆情况下做功,生成dA新表面所需环境做功(环境对系统做功时,系统得功,功为负值;系统对环境做功,系统失功,功为正值)为
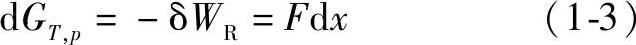
式中 δWR中的下标R——可逆;
G——表面吉布斯(Gibbs)自由能。
将式(1-1)代入式(1-3),得

于是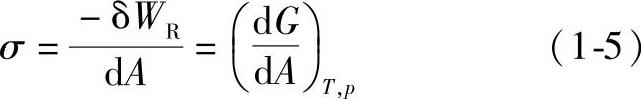
从热力学角度看,保持体系温度、压力、组成不变(封闭体系),增加单位表面积时吉布斯自由能的变化称为比表面吉布斯自由能。也可表示为等温等压、恒组成(封闭体系),增加单位面积所必须对系统做的非体积功,又称为比表面功,其单位为J·m-2。
对于液体来说,液体中原子或分子之间的相互作用力较弱,原子或分子的相对运动较易进行。液体内部原子或分子克服引力迁移到表面,形成新的表面,此时很快达到一种动平衡状态。一般认为,液体的比表面吉布斯自由能与表面张力在数值上是一致的。
3.固体表面及固体表面自由能
(1)固体表面的特点 固体表面具有不同于液体表面的一系列特性:
1)固体表面分子(或原子)移动困难。固体表面上的原子或分子与液体一样,受力也是不均匀的,所以固体表面也有表面张力和表面自由能。但固体分子或原子几乎不能自由移动,通常它们是定位的,因此固体表面不像液体那样易于缩小和变形。
2)固体表面的不完整性。根据组成固体的质点(原子、离子或分子)排列的有序程度,固体分为晶体和非晶体两类。非晶体中质点是杂乱无章的。在晶体中,质点以有序的空间晶格排列,由少数质点组成的重复单元组成。对于理想晶体,在温度一定时,晶胞大小和组成是相同的。但是,试验证明,几乎所有的晶体及其表面都会因为多种原因而呈现不完整性。即使同种晶体也会由于制备、加工不同而具有不同的表面性质,晶体表面的不完整性主要有晶格缺陷、空位和位错等。尽管这些不完整性比例很小,但对于表面吸附、表面催化、表面烧结都非常重要。
3)固体表面的不均匀性。固体表面是不均匀的,即使从宏观上看似乎很光滑,但从原子水平上看是凹凸不平的。正是由于固体表面原子受力不对称和表面结构不均匀,它才可以吸附气体或液体分子,使表面自由能下降。而且不同部位吸附和催化的活性不同。
(2)固体表面自由能 固体的表面自由能(固体比表面吉布斯自由能的简称),是指在某温度、压力下生成单位新固体表面所引起的体系能量的增加量。
与液体不同,固体表面张力不等于固体表面自由能。固体表面自由能等于固有的比表面自由能加上应力能,前者是单位面积的新表面所做的功,而后者是展开这个新表面所需要的功。为理解这两个概念的区别,对新表面形成的两步过程进行微观分析。
步骤一:首先,凝聚相(包括液相和固相)被分割易产生新表面,但新表面上的分子仍保留在本体相中所占据的确定位置(相对于残余的本体相)。
步骤二:接着,新表面中的原子重新定位到最稳定的构型。实际上,这意味着初生(新)表面上的某些单元被作用到它们上面的非平衡力拉入本体相中。
在液相体系中,由于单元的迁移性,使这两个步骤基本上同时发生。固体中原子或分子迁移率极大地降低意味着重排将十分缓慢,也许在一个合理时间尺度内根本不会发生。因此固体新表面上的单元密度也就不会达到平衡值。单元间的距离变化意味着它们的相互作用力以及吉布斯自由能会发生改变。固体新表面上的单元不能重新定位以达到与本体相关的平衡位置,受到表面应力的作用,因而这些表面分子具有应力能。
(3)表面应力 假设固体的表面沿垂直于表面的方向割裂开,新表面上的单元将不能重新定位以达到与本体相关的平衡位置。单位长度新表面达到平衡状态所需的力就是表面应力。固体与液体在形成新表面时有所不同。由于液体分子的可动性,形成新表面时,分子瞬间可以达到平衡,因此液体的表面自由能与表面张力相等。固体在形成新表面时,新生成固体表面上的分子也受到不平衡力的作用,它们应移动到平衡的位置上,但对于固体,这种移动很困难,需要很长时间才能完成。在未完成之前,这些分子受到应力τ的作用。使固体新表面上的分子(或原子)维持在未形成新表面前的位置上,单位长度所受到的力称为表面应力,或称为拉伸应力。若对两个互相垂直的割裂取平均表面张力,则将获得固体的表面张力。对于液体或各向同性的固体,两应力相同且表面应力与表面张力等值,即σs=τ1=τ2。而对于各向异性的固体,两者并不相同。固体的表面张力是新产生的两个固体表面的表面应力的平均值,即

式中 τ1和τ2——两个新表面的表面应力。固体表面张力不能像液体一样直接测出,实际处理问题时应用表面自由能会多一些。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。