



分子水平上,酶具有结合底物S和底物的活性位点。活性位点氨基酸残基和必需辅助因子通过共价和/或非共价键共同与底物发生相互作用。酶可以通过一系列机制催化键的形成/断裂和原子重排过程,这些作用受到活性位点上特有氨基酸残基及其空间排布的影响。除催化反应的必需氨基酸残基外,其他氨基酸残基也可以通过识别S来辅助催化反应。
(一)酶的活性中心
酶在催化反应中,起作用的并不是整个酶分子而是其中的某一部分。酶蛋白具有完整的空间结构,在肽链上存在许多侧链活性基团,如-NH2、-COOH、-OH、-SH等,某些侧链活性基团有可能随着酶蛋白的不同构象集合在一个特定的区域形成一定的空间构型。这个特定的区域就是酶蛋白分子中能与底物直接结合形成“酶-底物络合物”的特殊部位—酶的活性中心。活性中心的基团称为必需基团或活性基团。常见的必需基团有Ser-OH、Cys-SH、His-咪唑基、Asp-COOH、Glu-COOH、Lys-NH3等。根据其与底物作用时的功能不同,必需基团又可分成两种:一种是与反应底物结合的基团,称为结合基团;另一种是促进底物发生化学变化的基团,称为催化基团。
(二)酶-底物相互作用模型
Emil Fischer在1894年提出了酶-底物相互作用类似于锁和钥匙(Lock and key)的模型,Koshland在1959年提出了酶-底物相互作用的诱导契合(Induced-fit)模型。
按照锁和钥匙模型(图6-2),在酶的表面存在着一个特殊形状的活性部位,这个活性部位在结构上能与底物精确地互补。底物与酶之间存在某种立体专一性结合,底物类似钥匙,酶类似锁。该模型在葡萄糖氧化酶(Glucose oxidase EC 1.1.3.4)催化葡萄糖转化成葡萄糖酸的反应中得以验证。当采用结构上类似于葡萄糖的物质作为底物时,葡萄糖氧化酶的活力显著下降。例如,以2-脱氧-D-葡萄糖为底物时,葡萄糖氧化酶的活力仅为以葡萄糖为底物时的25%,以6-甲基-D-葡萄糖为底物时的活力不足以葡萄糖为底物时的2%,以木糖、半乳糖和纤维二糖为底物时的活力不足以葡萄糖为底物时的1%。这种假设能够解释酶的绝对特异性,但不能解释酶的相对特异性。然而许多酶催化反应不符合Fischer提出的锁和钥匙理论。
Koshland提出的诱导契合模型(图6-3)保留了在酶和底物之间形成络合物时具有立体选择性的概念,但是摒弃了酶的活性部位的结合部位是刻板结构并且即使没有底物存在时也保持精确无误的结构的概念。诱导契合模型的要点包括:①当底物与酶的活性部位结合时,酶蛋白的构象有相当大的改变;②催化基团的精确定向对于底物转变成产物是必需的;③底物诱导酶蛋白几何形状的改变使得催化基团能精确地走向和使底物结合到酶的活性部位上去。X-射线衍射分析的实验结果支持这一模型,证明酶与底物结合时,构象确实发生了变化。
图6-2 酶专一性的锁和钥匙机制
图6-3 酶与底物结合的诱导契合模型
很多实验数据都证明诱导契合概念比锁和钥匙的概念更加合适。诱导契合模型不仅可以解释符合锁和钥匙模型的实验结果,而且也能解释与经典理论相矛盾的结果。
(三)酶催化机制
根据过渡态理论,在任何一个化学反应体系中,反应物需要到达一个特定的高能状态以后才能发生反应。这种不稳定的高能状态被称为过渡态(激活态)。一个反应系统中各反应物分子具有不同的能量,只有某些反应物才含有足够的能量去进行反应。
1946年,鲍林(Pauling)用过渡态理论阐明了酶催化的实质,即酶之所以具有催化活力是因为它能特异性结合并稳定化学反应的过渡态(底物激活态),从而降低反应能级。Pauling提出,酶与底物过渡态的亲和力要比对底物基态的亲和力强得多,酶的催化源于其对底物过渡态的稳定作用。过渡态稳定是酶活性中心结构及反应性和活性中心与底物之间的相互作用的结果。酶充分使用一系列的化学机制来实现其对底物过渡态的结合与稳定并由此加速反应。这些机制归纳起来主要有四大类,分别是邻近效应、共价催化、酸碱催化、分子应变或扭曲效应(表6-5)。
表6-5 酶催化作用的一般机制
1.邻近效应
邻近效应的最好描述是:催化单元和底物以有利的方向挨近,从而促进反应的进行。另一种邻近效应的表述是:底物被定位于酶的活性部位,相对于溶液中的底物浓度,底物的“有效浓度”大大增加。因此,邻近效应对催化反应的贡献通常用有效(增加)浓度来建立模型,体现传质效应对反应速率的影响。
酶结合口袋使底物在酶的活性部位的低水环境中“对接”或“锚定”。底物与酶结合形成的复合物的寿命要比溶液中反应物分子碰撞产生的分子间结合的寿命长6个数量级。相互作用的寿命延长意味着使反应达到过渡态的可能性加大。因此,邻近效应对催化反应的贡献也可以将酶反应当作分子内反应来建立模型。相对于分子间反应,分子内反应的全部反应物被视为存在于单分子(酶分子)内。
邻近效应的净催化效应可以使1~3个底物参加的化学反应的速率提高104~105倍。
2.共价催化
共价催化是指反应过程中酶必须与底物上的某些基团暂时形成不稳定的酶-底物或辅助因子-底物共价中间物。该催化机制起始于亲核或亲电进攻。亲核中心富含电子(或具有非配对电子),有时候带负电,具有亲和羰基碳、磷酰基或糖基功能团等缺电子中心(核)并与之反应的倾向。亲核催化反应包括通过亲电体(也称电子穴,electron“sink”)获得反应中心的电子。当共价催化反应的反应物同时具有亲核和亲电基团时,根据首先启动反应的中心确定反应类型。
(1)亲核催化 亲核催化反应特有的步骤是亲核进攻。通常,亲核性与功能基团的碱度有关,碱度与能为质子提供电子对的能力有关。因此,亲核反应的速率常数与结构相关化合物的pKa呈正相关(pKa越大,反应速率越大)。然而,大多数情况下,酶的亲核基团只能在保持其构象稳定的pH范围内起作用。另一个影响亲核催化反应速率的因素是共价中间体形成过程生成的“离去基团”或产物的性质。离去基团的碱性越低(pKa越小),对同一亲核体来说亲核催化反应速率越快。
(2)亲电催化 亲电催化反应特有的步骤是亲电进攻。酶蛋白的氨基酸残基没有提供合适的亲电基团。亲电体来自缺电子的辅助因子或底物与酶的催化残基形成的带正电的含氮衍生物(表6-5)。一些最具典型的亲电催化反应以磷酸吡哆醛作为辅助因子。
3.广义酸碱催化
大多数酶反应涉及质子转移,通常是由可以提供质子(广义酸)和接受质子(广义碱)的氨基酸残基来完成的。能作为广义酸碱起作用的酶的氨基酸残基的pKa值通常在酶活和稳定性对应的最适pH范围内(一般为pH4~10,见表6-6)。广义酸碱行为对丝氨酸蛋白酶、脂肪酶以及羧酸酯酶的亲核作用机制也有贡献。组氨酸是参与广义酸碱催化反应较多的一个残基。
溶菌酶的最适pH在5附近,其催化反应机制取决于其活性部位的氨基酸谷氨酸Glu35和天冬氨酸Asp52的广义酸碱特性。Glu35质子起广义酸作用,与可断开的糖苷键的氧原子相偶合。如图6-4所示,天冬氨酸Asp52羧酸起广义碱作用,使底物变化中的正离子处于静电稳定。谷氨酸Glu35羧基失去H+后形成的COO-使完成水解反应所需要引入的水(未显示)电离,产生的OH-连接到原糖苷的C1上,糖苷键断裂。产生的H+被谷氨酸Glu35捕获结合,使酶回复到活性状态。
图6-4 溶菌酶的广义酸催化
广义酸碱催化通常可以使反应速率提高102~103倍。对溶菌酶而言,由于还存在其他对催化反应有利的因素(静电稳定和底物应变),总速率提高得更多。
4.应变和扭曲
与Fischer提出的“锁钥”概念不同,应变和扭曲机制是建立在底物和酶的作用域不是刚性的基础上的。Haldane和Pauling提出的扭曲或应变机制认为,当酶和底物结构互补而产生吸引力时,该种互补并不“完美”。如果“预先形成的”互补是“完美的”,催化反应发生的可能性不大,因为这样需要克服很大的能垒才能达到过渡态。
酶和底物结合位点预先形成的互补提供底物识别、结合能,同时帮助底物在活性部位上的定位。酶-底物结合产生结合能可以诱导酶和/或底物的应力/应变,实现互补的进一步“形成”。底物效应不大可能导致键的伸缩、扭曲或键角弯曲,因为要产生这些作用需要很大的力。一定程度上,键自由旋转受限制、空间压缩以及酶和底物静电排斥时,底物应变发生的可能性较大。因此,就真正的物理意义而言,底物结合到酶上时可能遭受“应力”(此时并不发生扭曲)。一些结合能的利用可以缓解应力,从而促进过渡态的形成。溶菌酶利用这种方式进行催化:底物中与溶菌酶活性中心结合的六碳糖(吡喃糖衍生物)在溶菌酶的诱导下从较稳定的椅式构型变成相对更不稳定的半椅式构型,使得周围的糖苷键更容易发生断裂。
除上述归纳的四种酶催化作用机制外,还有金属离子催化和静电催化。金属催化是指金属离子参与的催化。近三分之一已知酶的活性需要金属离子的存在。静电催化是指酶使用自身带电基团去中和一个反应过渡态形成时产生的相反电荷而进行的催化。有时,酶通过与底物的静电作用将底物引入到活性中心。
由于酶分子是一个由多种不同侧链基团组成活性中心的大分子,这些基团在催化过程中根据各自的特点发挥作用,因此在酶催化反应中,常常是几个基元催化反应配合在一起的共同作用,其作用效率远远胜过单个基元催化(广义酸碱催化、共价催化以及金属离子的催化)的效率。相对于非催化反应,在酶催化反应中,上述不同作用机制总的可以使反应速率提高1017~1019倍。
食品加工的生物材料中含有数以百计的不同种类的酶,这些酶对于原料的生长、成熟、加工和保藏等均起着重要的作用。例如,苹果、香蕉、葡萄、洋李、茶叶和咖啡豆等含有大量的多酚氧化酶,大豆含有丰富的脂肪氧合酶,青刀豆、卷心菜等含有活性很高的过氧化物酶,番茄、柑橘、牛油果等含有大量的果胶酶等。在现代食品加工保藏中,为达到某个加工或保藏的目的经常有目的地使用外源酶。另外,食品原料、中间品和产品污染的微生物也会分泌产生酶。不管是内源酶还是外源酶,对食品的质量都有重要影响,因此在原料处理、加工和食品保藏过程中必须对它们的活力加以控制。下面就影响酶催化反应的主要环境因素进行讨论。
(一)pH
pH对大多数酶的活力都有显著影响。若以酶的活力或反应速度对pH作图,可以得到一个pH -活力曲线(图6-5和图6-6)。处于曲线最高点处的pH称为最适pH,在最适pH处酶的活力最大。每一种酶都有其最适pH(图6-5),这是酶的一个重要特征。缓冲液的种类对酶的最适pH也有影响。例如,脲酶对尿素的催化反应,由于缓冲液的不同,脲酶显示不同的最适pH(图6-6)。
图6-5 pH对几种酶活力的影响
1—胃蛋白酶作用于N-乙酰-L-苯丙氨酰-L-二碘酪氨酸
2—过氧化物酶作用于愈创木酚
3—胰凝乳蛋白酶作用于酪蛋白
4—碱性磷酸酯酶作用于对硝基苯磷酸酯
图6-6 缓冲液种类及pH对脲酶活力的影响
1—乙酸盐 2—柠檬酸盐 3—磷酸盐
注:脲的浓度为2.5%
pH对酶催化反应影响的原因是多方面的。一方面,过酸或过碱可能改变酶的空间构象,使酶失活,根据程度不同,酶可遭受可逆或不可逆失活。另一方面,pH可改变底物的解离状态,影响底物与酶的结合,从而影响酶催化的效率。酶反应介质的pH可影响酶分子,特别是活性中心上必需基团的解离程度和催化基团中质子供体或质子受体所需的离子化状态,也可影响底物和辅酶的解离程度,从而影响酶与底物的结合。只有在特定的pH条件下,酶、底物和辅酶的解离情况最适宜于它们互相结合,并发生催化作用,使酶促反应速率达最大值。常见于食品材料的几种酶最适pH列于表6-6中。最适pH与酶的来源和实验条件有关,表中所列的最适pH应是实际近似值。
表6-6 一些酶的最适pH
续表
酶不是在所有的pH下都稳定,而是有一个稳定的pH范围。酶的最适pH和酶的最稳定pH不一定相同。
确定酶的pH稳定性的最佳方法是在与测定酶催化反应初速度相同的条件(温度、缓冲液和酶浓度等)和不同pH下将酶保温,在不同的时间取出一定量的酶液加入到含有底物(已被缓冲至酶的最适pH或接近酶的最适pH)的试管中,测定酶催化反应的初速度。用曲线表示酶在不同的pH下保温时残存的酶活力随保温时间的变化(图6-7)。
图6-7 酶在不同的pH下保温时的失活速度
注:以未经保温处理的酶样的活力为100,计算百分数残余活力
如果pH仅影响酶的变性,那么酶的残余百分活力的对数-保温时间关系曲线是一条直线,直线的斜率是酶失活的一级速度常数。
酶的pH稳定性数据对于确定酶的保藏和使用条件都是重要的。虽然在实验室中往往是通过测定酶催化反应的初速度和pH的关系来确定酶的最适pH的,然而在食品加工中酶作用的时间相当长,因此在确定应采用的pH时,除了考虑酶的最适pH外,还要考虑酶的pH稳定范围。
(二)温度
温度是影响酶活力的另一个重要参数。在一定温度范围内,酶催化反应的速度随着温度的升高而升高,是因为温度影响酶催化反应中ES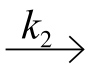 E+P的速度,这一步的速度常数k2=
E+P的速度,这一步的速度常数k2=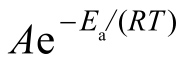 。当温度超过某一值时,酶反应速度随温度的升高而下降,这是因为酶的热变性使酶的活力下降。另外,酶反应中所有的缔合/离解平衡(缓冲剂、底物、产物和辅助因子离子化)、酶-底物络合物的缔合/离解、可逆酶反应(S
。当温度超过某一值时,酶反应速度随温度的升高而下降,这是因为酶的热变性使酶的活力下降。另外,酶反应中所有的缔合/离解平衡(缓冲剂、底物、产物和辅助因子离子化)、酶-底物络合物的缔合/离解、可逆酶反应(S P)、底物(尤其是气体)的溶解度、酶活性部位、酶-底物络合物中的质子移变基团的离子化等都与温度有关。
P)、底物(尤其是气体)的溶解度、酶活性部位、酶-底物络合物中的质子移变基团的离子化等都与温度有关。
1.酶的热稳定性
酶的热稳定性可按下述方法测定:在一组试管中加入固定的酶量和相同的缓冲液,缓冲液的pH必须调节到期望的数值(酶在此pH下是稳定的),将试管在不同的温度下保温,一般从25℃开始,温度间隔为5℃。在不同的时间从各个试管取一定量的酶液测定残余酶活力,测定酶活力所采用的底物浓度、缓冲液pH和温度必须保证酶处在最适条件。将保持在0℃的酶样的活力作为对照活力(100%),计算在各个不同温度和不同保温时间酶的残余活力。由于酶的热失活通常遵循一级动力学,因此酶的残余活力百分数的对数与保温时间呈线性关系(图6-8),直线的斜率k是酶的变性速度常数。如以酶热失活速度常数k的对数lgk对1/T(K)作图,可以得到一条直线。根据Arrnenius方程式 ,直线的斜率为-Ea/2.3R,Ea是酶热变性(失活)的活化能,R是通用气体常数。
,直线的斜率为-Ea/2.3R,Ea是酶热变性(失活)的活化能,R是通用气体常数。
图6-8 温度对酶稳定性的影响
图6-9 温度对酶催化反应产物形成的速度的影响(实线是实验数据,虚线是根据初速度外延得到的)
图6-10 温度对酶催化反应速度常数的影响
2.酶催化反应的活化能
测定酶催化反应的活化能需要作两种图。第一个图是在不同温度下实验测定的反应产物浓度-反应时间图(图6-9),根据此图计算不同温度下酶催化反应的速度常数k。第二个图是酶催化反应的速度常数k的对数lgk对1/T(K)图(图6-10),从直线的斜率计算酶反应的活化能Ea(斜率=-Ea/2.3R)。对于第一个图所选择的温度必须保证所测定的初速度没有受到酶热变性的影响。同时,在所有的温度条件下酶必须被底物饱和,即保证[S]0≥Km。另外,采用相同的pH,而且必须是最适pH。
由 可以看到,提高温度对反应速度的影响取决于活化能Ea。高活化能意味着温度对反应速度的影响大,即反应速度随温度的升高提高很快。酶通常能降低反应的活化能,因此酶会产生两个效果:在低温下使较高比例的反应物转变成产物;升高温度对酶催化反应速度造成的影响相对较小。然而,即使对一个活化能为50kJ/mol的酶催化反应,当温度从22℃升高至32℃时,反应速度仍然能提高至原来的2倍。因此,在酶稳定的温度范围内,尽可能地采用高温仍然是有益的。(https://www.xing528.com)
可以看到,提高温度对反应速度的影响取决于活化能Ea。高活化能意味着温度对反应速度的影响大,即反应速度随温度的升高提高很快。酶通常能降低反应的活化能,因此酶会产生两个效果:在低温下使较高比例的反应物转变成产物;升高温度对酶催化反应速度造成的影响相对较小。然而,即使对一个活化能为50kJ/mol的酶催化反应,当温度从22℃升高至32℃时,反应速度仍然能提高至原来的2倍。因此,在酶稳定的温度范围内,尽可能地采用高温仍然是有益的。(https://www.xing528.com)
3.酶变性(失活)的活化能
酶热变性(失活)的活化能随酶的种类而异,通常在167~418kJ/mol范围内。各种酶的热稳定性差别很大:聚半乳糖醛酸酶和胰蛋白酶在60℃时仍具有活性,而葡萄糖磷酸异构酶在比60℃低很多的温度条件下就失活。
4.低温时酶的活力
溶液或食品冻结以后,酶的活力并没有完全停止。图6-11所示为温度(49.6~19.4℃)对转化酶催化蔗糖水解和温度(25~60.2℃)对β-半乳糖苷酶催化o-和p-硝基苯-β-半乳糖苷水解的影响。从图6-11可以看出,即使转化酶和β-半乳糖苷酶的活力分别下降105和104.3倍,它们在整个研究的温度范围仍然具有活力。在-3℃,A线的斜率发生变化,且在此温度下溶液开始冻结。对斜率的变化可以作如下解释:①在溶液的冻结点发生了相变;②限制速度的反应步骤改变;③酶缔合成聚合物;④底物与水的氢键增加。为了防止冻结,β-半乳糖苷酶催化o-和p-硝基苯-β-半乳糖苷水解的反应在50%的二甲基亚砜中进行,图6-11的B和C线的斜率没有改变。在25~60.2℃的温度范围只要不出现冻结现象,就可以根据Arrhenius方程式预测β-半乳糖苷酶的活力。
图6-11 温度(49.6~60.2℃)对酶催化反应速度的影响
A●转化酶催化蔗糖在缓冲液中的水解,两条虚线的交点表明酶催化反应速度的变化在溶液的冻结点出现了转折
B□β-半乳糖苷酶催化o-硝基苯-β-半乳糖苷的水解,pH6.1,50%二甲基亚砜-水
C△β-半乳糖苷酶催化p-硝基苯-β-半乳糖苷的水解,pH7.6,50%二甲基亚砜-水
食品应避免被贮存在冰点或刚低于冰点的温度。当水冻结时,酶和底物被浓缩(溶质不存在于冰相),会促进酶的活力。此外,冻结和解冻会破坏食品的组织结构,使酶更易接近底物。如图6-12所示,鳕鱼肌肉中磷脂酶的活力在-4℃(低于冷冻点)时的活力相当于-2.5℃时的5倍。
5.酶反应的最适温度
酶的最适温度是指酶反应速度最大时所对应的温度。酶反应的最适温度可以根据温度对酶的净激活效应和净失活效应得到。图6-13(1)所示为温度对产气杆菌普鲁兰酶活性的影响。在10~40℃时,酶活曲线缓慢上升;在50~60℃时,失活速度常数迅速增加,酶活和稳定性迅速下降。图6-13(2)所示为一些与食品相关酶的活力与温度的关系和稳定温度范围。酶的最适温度是操作参数。在实际应用时,食品体系中的酶反应温度上限通常要比酶反应的最适温度低5~20℃,从而使酶活在规定的处理时间内保持较高水平,得到较多的产物。因此,可以说,实际应用的酶的最适温度是指在规定的时间内得到最多产物的温度,是与反应时间有关的操作参数。设定的反应时间越长,最适温度越低。
图6-12在冰点以下温度鳕鱼肌肉中磷脂酶催化磷脂水解的速度常数(k)
图6-13 温度对一些酶活性的影响
(三)水分活度
控制食品含水量是食品保藏的主要方法,几乎所有的食品都有特定的水分含量范围。水作为酶反应的分散介质,通过控制溶质的稀释度或浓度、稳定化和塑化蛋白质和作为水解反应的共底物来影响酶反应速率。脱水或冷冻会减少体相(或溶剂)水,导致食品组成和物性改变,从而对酶反应产生影响。
酶通常在含水的体系中作用。图6-14所示为水分活度对磷脂酶和β-淀粉酶活力的影响。水分活度低于0.35(<1%水分含量)时,磷脂酶没有水解卵磷脂的活力,超过0.35时,酶活力非线性增加,在0.9时仍然没有达到最高活力。在水分活度高于0.8(2%水分含量)时,β-淀粉酶才显示出水解淀粉的活力,当水分活度达到0.95时,酶的活力提高15倍。从这些例子可以得出结论:食品原料中的水分含量必须低于1%~2%才能抑制酶的活力。
图6-14 水分活度对酶活力的影响
可以采用有机溶剂取代水分的方法来确定酶作用所需的水的浓度,例如,采用能与水相混溶的甘油取代水分。当水分含量减少至75%时(图6-15),脂肪氧合酶和过氧化物酶的活力开始降低;当水分含量分别减少至20%和10%时,脂肪氧合酶和过氧化物酶的活力降至0。需要注意的是,黏度和甘油的特殊效应可能会影响到这些结果。
图6-15 甘油在水中的浓度对过氧化物酶(▲)和脂肪氧合酶(●)催化反应的速度的影响
图6-16 水分活度对不同来源脂酶的酯合成活力的影响
图6-16是水分活度对不同脂酶的酯合成活力的影响。在酯合成反应中,不同来源的脂酶具有不同的特定的最适水分活度。
不同酶发挥催化功能所需的最低的水分活度不同。当水分活度处于或低于单层水的水分活度时,酶分子的塑性受到限制,但仍然有一些酶表现活性。低于单层水时的水分活度抑制酶的反应活性,但却能提高酶的热稳定性,因为其构象变化同样受到限制,在变性温度下也不容易变性。在食品基质和模型系统中一些酶对水分活度的要求见表6-7。一些氧化-还原酶类所需的最低水分活度范围为0.25~0.70,一些水解酶类所需的最低水分活度范围为0.025~0.96。中等含水量食品在长时间贮藏过程中,即使很低的残余酶活也会影响食品品质。
表6-7 一些酶对水分活度的要求
降低水分活度的另一个效果是影响水参加反应(水解反应)的平衡。当水分活度下降时,反应朝着合成方向移动。利用这一原理进行产品合成或改性已有成功实例。例如,利用脂酶在微水相介质(H2O<1%)中生产功能得到改善的脂类产品;利用嗜热菌蛋白酶在微水相介质(H2O含量2%~3%)中合成阿斯巴甜。
有机溶剂对酶催化反应的影响主要有两个方面:影响酶的稳定性和反应(反应是可逆的)进行的方向。这些影响在与水不能互溶和能互溶的有机溶剂中是不同的。在与水不能互溶的溶剂中,酶的专一性从催化水解反应移向催化合成反应。例如,当“干”(1%水分含量)脂酶颗粒在与水不能互溶的有机溶剂中悬浮时,脂酶催化脂的酯交换反应的速度提高6倍以上,而水解速度下降16倍。
酶在水-与水能互溶溶剂体系中的稳定性和催化活力不同于在水-与水不能互溶溶剂体系中的情况。蛋白酶催化酪蛋白水解在5%乙醇-95%缓冲液或5%丙腈-95%缓冲液中进行时与在缓冲体系中进行时相比Km提高,vmax降低和酶稳定性下降。在水解酶催化的反应中醇和胺与水存在着竞争作用。
(四)电解质
对于一些酶,某些离子对其催化活力是必须的。一些酶催化反应必须有其他适当物质存在时才能表现出催化活力或加强其催化效力,这种作用被称为酶激活作用,引起激活作用的物质称为激活剂。激活剂和辅酶或辅基不同。无激活剂存在时,酶仍能表现一定的活力,而辅酶或辅基不存在时,酶则完全不呈现活力。
激活剂的种类很多,有无机阳离子,如:Na+、K+、Rb+、Cs+、 、Mg2+、Ca2+、Zn2+、Cd2+、Cr3+、Mn2+、Fe2+、Co2+、Ni2+、Al3+等;有无机阴离子,如:Cl-、Br-、I-、CN-、
、Mg2+、Ca2+、Zn2+、Cd2+、Cr3+、Mn2+、Fe2+、Co2+、Ni2+、Al3+等;有无机阴离子,如:Cl-、Br-、I-、CN-、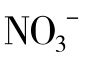 、
、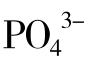 、
、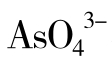 、
、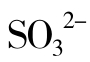 、
、 、
、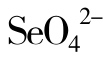 等;有有机物分子如维生素C、还原型谷胱甘肽以及维生素B1、维生素B2和维生素B6的磷酸酯等化合物和一些酶。
等;有有机物分子如维生素C、还原型谷胱甘肽以及维生素B1、维生素B2和维生素B6的磷酸酯等化合物和一些酶。
一般认为,金属离子的激活作用是由于金属离子与酶结合,此结合物又与底物结合形成三位一体的“酶-金属-底物”的复合物,这里金属离子使底物更有利于同酶的活性部位相结合而使反应加速进行。金属离子在其中起到了某种“搭桥”的作用。
无机阴离子对酶的激活作用也是常见的现象。例如,Cl-是唾液淀粉酶活力所必需的,当用透析法去掉Cl-时,淀粉酶便丧失其活力。无机阴离子的激活机制目前还不太清楚,有人认为这里的阴离子是酶的活力所必需的因子,而且对酶的热稳定性也起保护作用。
有些物质能抑制甚至阻止酶催化反应的进行。这种作用称为酶的抑制作用,这种物质称为抑制剂。抑制剂的种类繁多,如无机物中的强酸、强碱、重金属离子(Hg2+、Pb2+或Ag+)、一氧化碳、硫化氢等,有机化合物中某些碱、染料、EDTA、SDS、脲或酶催化反应的自身产物等。抑制作用有可逆性抑制和不可逆性抑制。抑制剂一旦与酶结合,再也不能用透析或稀释等方法将其除去,酶的催化活力永久性丧失,称为不可逆抑制。不可逆抑制一般都是抑制剂与酶的活性中心基团结合,从而使酶活性中心的必需基团改变或构象改变。例如,重金属离子、碘乙酸等与巯基酶活性中心的半胱氨酸残基的-SH结合,有机磷化物能与多种酶活性中心的丝氨酸残基上的-OH结合。这些结合都不能通过稀释、透析等方法除去抑制剂,所以,引起的酶活力的丧失不能恢复。可逆抑制是指抑制剂能快速地(在几个毫秒内)与酶形成非共价扩散控制平衡络合物,但通过透析或凝胶过滤色谱可使此平衡络合物解离和酶活力复原。酶的可逆抑制及其动力学将在后面专门讨论。
(五)剪切作用
在食品加工过程中的一些操作,例如混合、管道输送和挤压会产生明显的剪切作用。导致酶失活的剪切条件与酶的性质有关。图6-17阐明了在4℃时因剪切作用所造成的凝乳酶的失活。当剪切值[剪切速度(s-1)乘以剪切时间(s)]大于104时就能检测到酶失活;当剪切值达到107数量级时,过氧化氢酶、凝乳酶和羧肽酶的50%左右活力会丧失。不过,凝乳酶在剪切失活后,当剪切作用停止后又会部分再生。这一现象类似于酶热失活后的部分再生。
图6-17在4℃时剪切作用使凝乳酶失活
(六)压力
在通常的食品加工过程中,所采用的压力一般不至于高到使酶失活。然而在采用几种处理方式相结合的加工过程中,例如压力-高温处理和压力-高剪切处理,都能导致酶失活。挤压机在高于食品加工中通常使用的压力下操作时,由于高剪切而造成食品更高的流动性、组织化、化学变化和高温。显然,在这样的加工条件下食品原料中大多数酶难以再存活。在单独使用静水压力的情况下,只要食品组织还能保持完整,那么食品原料中的酶就不会完全失去活性。不过,近年来超高压(400~1000MPa)技术在食品加工与保藏中的应用越来越多,数百兆帕的静水压力对大多数酶的活力有显著的影响。
压力对酶活力的影响还与酶的结构有关。对于多亚基酶,高压会导致酶蛋白解离成单体,不利于酶的稳定和催化活力。
(七)电场
外加电场会影响蛋白质内部基团间的静电相互作用和带电基团的定位,扰乱蛋白质氨基酸残基间的电场分布和静电相互作用,导致电荷分离,从而影响蛋白质的空间结构。脉冲强电场还可以与蛋白质分子结构单元(如α-螺旋)肽键偶极矩叠加产生的定向偶极矩发生耦合,从而影响蛋白质二级结构。已有许多研究表明高压电场对模拟体系或真实食品体系中的酶有活化和钝化两种情况。
一般来说,无论以α-螺旋还是β-折叠为主的酶在高压电场作用下的钝化效果都与其二级结构的改变程度有关,并且钝化效果也与电场强度、酶的种类与溶液的性质有关。此外也有研究表明电场作用会改变酶的活性区域结构,从而影响酶与底物结合而降低酶活。
当电场强度在一定范围内对酶有活化作用,其可能是通过电场作用激发了更多的酶活性位点或者是增大了酶现存活性位点起催化作用的范围,从而使酶的活性得到提高。同样地,电场对酶的活化效果与酶的种类、溶液性质与电场作用条件相关。近三十年来,有关高压脉冲电场对酶蛋白的结构与活性及其他蛋白质的结构与功能的影响的研究报道比较多。
(八)超声波
采用超声波处理食品体系时产生的空化作用会导致酶蛋白的界面变性,这个观点在研究木瓜蛋白酶时得到了证实。木瓜蛋白酶因超声波作用而失活的程度反比于酶的浓度,酶失活过程不符合一级动力学。
(九)离子辐射
离子辐射能使酶失活,但是离子辐射使酶完全失活所需的剂量比破坏微生物所需的剂量大10倍。酶的性质、水分活度、酶的浓度、酶的纯度、在体外还是在体内、氧浓度、pH和温度等都会影响离子辐射对酶的破坏作用。在缺氧和干燥状态下,大多数酶对辐射引起的失活具有大致相同的抵抗力。但是,处于稀溶液中的不同的酶对辐射的敏感程度有很大差异。如图6-18所示,胰蛋白酶在干燥状态时抵抗辐射损害的能力很强,这主要是在此状态下起作用的仅仅是辐射的直接效应;当有水存在时,胰蛋白酶对辐射比较敏感,这可能是在有水时“间接效应”变得重要。间接效应包括水的辐解和生成的自由基的损害效应。
一般来说,在一定的限度内,酶浓度愈稀,破坏相同百分比的最初酶活力所需的剂量就愈小,此现象被称为稀释效应;纯度较低的酶制剂通常比纯的酶制剂更能抵抗辐射损害;氧往往使酶对辐射更敏感,这或许是由于氧的存在会导致不稳定的中间物-蛋白质的过氧化物的形成;温度的影响很明显,当温度较高时,酶很容易辐射失活,但对于冷冻体系,辐射对酶的损害很小。据推测,这是由于在冷冻体系中自由基是固定化的;pH对酶对辐射的敏感度的影响也值得重视,但是这种影响很难被预测。
图6-18 各种条件下辐射导致胰蛋白酶失活
1—干胰蛋白酶
2—10mg/mL浓度的胰蛋白酶溶液、pH8和-78℃
3—80mg/mL浓度的胰蛋白酶溶液、pH8和室温
4—10mg/mL浓度的胰蛋白酶溶液、pH2.6和-78℃
5—80mg/mL浓度的胰蛋白酶溶液、pH2.7和室温
6—10mg/mL浓度的胰蛋白酶溶液、pH2.6和室温
7—1mg/mL浓度的胰蛋白酶溶液、pH2.6和室温
(十)界面作用
蛋白质一般倾向于被吸附在界面,这往往会造成蛋白质变性。其原因显然是在气/水或油/水界面上具有高的界面张力,在界面上酶的二级和三级结构很快的展开,并且在整个界面上铺展开来。在气/水界面,疏水氨基酸残基指向空气而亲水氨基酸指向水;在油/水界面,疏水残基同油相缔结而亲水侧链伸展到水中。
使酶溶液简单地起泡就能产生导致蛋白质表面变性的气-水界面。在各种泡沫中酶活力的损失不易通过压缩或回收膜的方法来重新获得。在油/水界面上的吸附效应类似于在气-水界面上的吸附效应。然而,当酶吸附在已吸附了大量的另一个蛋白质或“保护”分子的界面时,不会再出现酶蛋白分子展开和酶活力损失的现象。
脂酶是一种特别适用于在油/水界面上作用的酶,但是当被吸附在甘油三酯水界面上时,仍然会发生变性。脂酶在气/水界面上也会失活。
(十一)溶剂作用
溶剂会影响体系的水分活度,从而影响酶的催化活性。一般地讲,与水不能互溶的溶剂通过置换水而稳定酶,与通过干燥将水除去所产生的效果一样。然而,与水能互溶的溶剂在浓度超5%~10%时,一般会使酶失活。显然,此效应取决于温度(在较低温度下酶较稳定)。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。