



材料特性是电子封装领域的一个热门研究主题,尤其研究电子封装大量使用的交联环氧树脂混合物。为了在电子封装中设计和研发交联环氧树脂,预测和准确理解交联环氧树脂的特性就变得必要且重要。分子动力学模拟已经被用来预测不同材料的材料特性[53-57]。而这些研究的大部分都集中在热塑性材料领域,如聚乙烯(甲基丙烯酸甲酯)、聚乙烯和酚酞聚芳醚砜等。但是,由于交联反应的复杂性,对热固性材料的材料特性的研究少之又少,尤其是在电子封装中广泛应用的交联(Crosslinked)环氧树脂复合材料(Epoxy Resin Compound,ERC)。Fan等人[25]采用分子动力学模拟技术研究了交联环氧树脂混合物的材料特性,具体如下。
在其研究中,所有的分子动力学模拟中均采用聚合物调和力场(Polymer Con-sistent Force Field,PCFF)进行,并采用速度蛙跳算法来计算积分,采用由范德华力和静电场力组成的非键合作用力和截断距离为9.5Å(1Å=0.1nm)的Ewald加和来计算散射作用。充分固化的环氧树脂网状物含有环氧树脂EPON862和固化剂三乙烯四胺(Triethylenetetramine,TETA),其中单体EPON862和TETA的化学结构如图3.7所示。在固化反应中,固化剂中胺基团上的氢原子与环氧树脂中的环氧基团进行反应,这种交联反应向各个方向扩展,并最终形成高分子网状物。
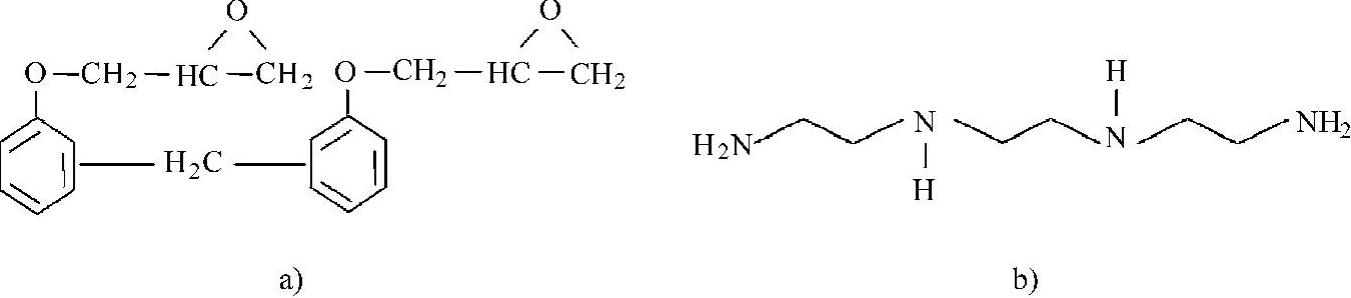
图3.7 EPON 862和TETA的化学结构
a)EPON 862 b)TETA
基于参考文献[58]的实验条件,我们建立了固化环氧树脂的网络片段模型。它含有12个EPON 862分子和4个TETA分子,同时也构造了一个密度等于实验值1.23g/cm3的无定形结构,且模型中的模拟单元在各方向上均呈周期性分布。并且,采用起始温度是225℃、压力为0.1MPa的等温等压系综来进行分子动力学模拟,还采用nose温控方法以10℃/200ps的冷却速率将系统冷却到室温。而后续每次分子动力学模拟的系统初始状态都是上一次模拟温度条件下所得到的最终系统结构。并且,一次分子动力学模拟中,每个模拟步骤的时间间隔是1fs[2]。
图3.8a给出了每个温度下固化环氧树脂的密度值。模拟结果表明,随着温度的下降,其密度稳定地增大,而其密度曲线的斜率则变小。其密度曲线的斜率发生变化时的温度即为固化环氧树脂的玻璃态转变温度(Tg),接近109℃。通过分子动力学模拟得到的Tg值非常接近表3.2中列出的、参考文献[58]提供的实验值。
表3.2 经分子动力学模拟和实验获得的环氧树脂复合材料的材料特性数据[58]

固化环氧树脂系统的初始体积为36.20nm3,而每个温度下系统的相应体积都是通过分子动力学模拟计算得到。图3.8b所示为单元结构的体积随温度变化的关系。利用其密度曲线在玻璃态转变温度前后的斜率值,可以分别得到橡胶态和玻璃态下环氧树脂复合材料的体积热膨胀系数。线性热膨胀系数与体积热膨胀系数之间的关系如下:(https://www.xing528.com)

玻璃态和橡胶态下环氧树脂复合材料的线性热膨胀系数见表3.2。
等效连续本构关系能描述原子级系统的力学行为。对于各向同性材料,与宏观系统一样,原子级系统的应力-应变关系可仅通过两个独立的系数λ和μ,即Lame系数来描述,因此分子动力学模拟中材料的杨氏模量可定义为

表3.2也给出了分子动力学模拟所得的环氧树脂复合材料的杨氏模量。从该表可知,分子动力学模拟所得的热膨胀系数和杨氏模量与参考文献[58]给出的实验结果非常接近,误差小于10%。两种结果之间的微小误差是由实验情况下[58]环氧树脂复合材料的交联密度较低所致。在实验情况下,部分的EPON 862和TETA不会充分交联在一起,而分子动力学模拟中EPON 862和TETA则充分交联,故与模拟所用的模型相比实验样品会产生更大的应变。因此,实验情况下的环氧树脂复合材料更软,而且要承受更大的形变,从而导致其杨氏模量和热膨胀系数降低。此外,实验情况下环氧树脂复合材料内部会存在空洞和杂质,这也进一步降低了其材料特性[25]。
分子动力学模拟预测所得的材料特性与参考文献[58]给出的实验结果非常接近,这证实了固化环氧树脂模型及力场特性和冷却速率的准确性。因此,在具体实验室设计之前,通过分子动力学模拟对环氧树脂复合材料进行性能预测,能有效地指导电子封装领域内新型高性能环氧树脂复合材料的设计和开发。
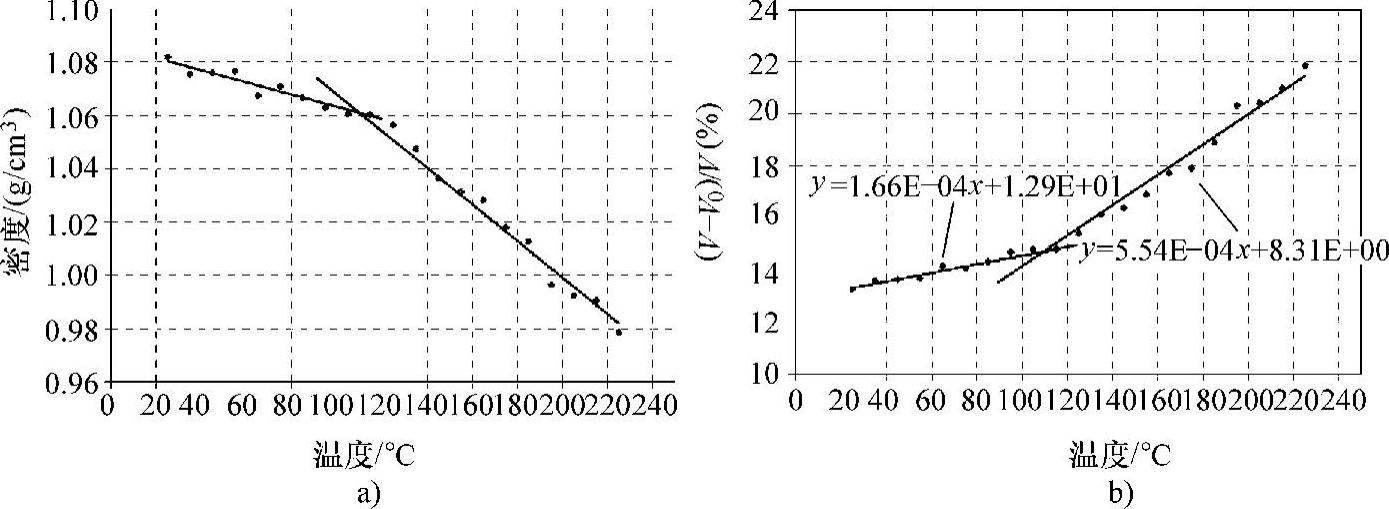
图3.8 密度与温度的关系、体积与温度的关系及各自的拟合直线
a)密度与温度的关系 b)体积与温度的关系
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。