



毛细管流变仪黏性流体的流场分布不同于牛顿流体,一定要修正测试的流变数据。本小节重点分析毛细管熔体入口压力损失和测量数据的修正,包括入口压力损失、Bagley修正、入口压力降的应用实例、双毛细管流变仪4部分。
分析毛细管入口区的流场。物料在毛细管管壁处承受的剪切应力τw是通过测量完全发展流动区上的压力梯度∂p/∂z确定的,有
当压力梯度均匀时,计算压力梯度的简便公式为
式中,Δp为长度为L完全发展流动区两端的压力差。
但是,在实际使用毛细管流变仪测量物料流变性能时测量时,由图8.2.1和图8.2.4均可见,压力传感器安装的位置不在毛细管上,而是在料筒壁处。通过控制活塞下压速度来控制剪切速率,用压力传感器测量压力,测量压力计算剪切应力。由于物料从料桶被挤入到毛细管口模时,各个流层之间产生黏性摩擦,从而消耗了能量;还有物料的弹性形变,毛细管入口压力传感器测得的压力差是入口、完全发展区和出口压力降的总和,如图8.2.4(a)和(b)所示。
测量压力差的表达为
另外,完全发展流动区的流道长度与毛细管长也不相等。因此在通过测压力差来计算压力梯度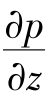 时,必须进行适当的修正。
时,必须进行适当的修正。
由图8.2.5料筒与毛细管中物料内部压力分布示意图可看出,对于黏弹性流体,当从料筒进入毛细管时,存在着一个很大的入口压力损失Δpent。这个压力损失是黏弹性流体流经截面形状变化流道时的重要特点之一,由于物料在入口区经历了强烈的拉伸流动和剪切流动,以至于贮存和损耗了部分能量。
实验发现,在全部入口压力损失中,95%是由于弹性能储存引起的,仅5%是由于黏性损耗引起的。对于纯黏性的牛顿型流体而言,入口压力降很小,可忽略不计。而对黏弹性流体,则必须考虑因其弹性变形而导致的压力损失。
图8.2.4 毛细管流变仪压力传感器测量压力的示意图
(a)仪器示意(b)压力分布
相对而言,出口压力降比入口压力降小得多。对牛顿型流体来讲,出口压力降为零,等于大气压。对于黏弹性流体,若其在毛细管入口区的弹性变形在经过毛细管后尚未全部松弛,至出口处仍残存部分内压力,则将表现为出口压力降Δpexit。在本节研究毛细管上压力分布时,暂不考虑出口压力降的影响。第8.2.4节分析出口区流体的流动。
图8.2.5 料筒与毛细管内物料内部压力分布示意图[5]
为了从测得的压力差Δp准确地求出完全发展流动区上的压力梯度∂p/∂z,Bagley提出一种修正方法。在文献[2,4,6]中都详细介绍了Bagley修正。将毛细管的完全发展流动区虚拟地延长,保持压力梯度∂p/∂z不变,假设入口区压力降等价为在虚拟延长长度上的压力降。
Schramm著作中介绍了Bagley的修正方法[2,3]。用几个同直径毛细管口模(至少两个)对被测聚合物做若干实验,其他实验条件相同,只是毛细管的长/径比(L/D)不同,如10、30和40。因为,毛细管口模入口区相同,用这些毛细管所做的实验显然具有同样的入口效应。毛细管口模的L/D的比越小,入口效应产生偏差的百分比越大。可以由大的长径比的口模外推至小长径比的其他口模,做此外推,得到最终的结论:
假设一种极端情况,口模长为零或L/D=0,测得的压力差刚好是由总入口效应所产生,正是力求测得的数据。
因此,用几个直径相同,不同长径比口模做实验,设定剪切速率值,绘制压力降随口模长径比的变化曲线Δp-L/D,如图8.2.6所示是一组呈扇形的直线。将这些直线延伸,交于纵坐标于Δpc点,下标c代表修正。Δpc即为口模长L=0的毛细管的入口效应所产生的应力降。也可将上述直线继续延伸,与横坐标相交,得到负的L/D(绝对值计为nB)或ΔL(ΔL=nBD)。用Δpc和ΔL都可进行Bagley修正。综上所述,得到Bagley两种修正的方法:
① 将测定压力降Δp减去Δpc,有剪切应力的修正值为
② 将毛细管长度L加上长度ΔL,有剪切应力的修正值为
图8.2.6 确定棒状毛细管口模入口效应的Bagley修正因子[3]
由于入口压力降主要因流体存储弹性能引起,因此一切影响材料弹性的因素,例如相对分子质量、相对分子质量分布、剪切速率、温度、填料等都将对nB值产生影响。实验发现,当毛细管长径比L/D较小,而剪切速率较大,温度较低时,入口修正不可忽视,否则不能求得可靠的结果。手工进行棒状毛细管的Bagley修正即费时又麻烦。可以使用计算机软件在很短时间完成该修正。(https://www.xing528.com)
为了工程上使用方便,Macosko[4]给出表观剪切速率和剪切速率的近似关系,有
流量和压力梯度的关系为
可以用表观黏度近似公式计算被测流体的黏度,有
参考文献[2]给出非牛顿流体用表观黏度近似计算被测流体黏度的经验公式,对于中等剪切速率的聚合物熔体,可用幂律公式拟合非牛顿流体的黏度
对于牛顿流体,若n=1,上式简化为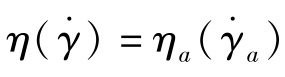 。
。
对于非牛顿流体,则有
介绍用入口压力降表征硬聚氯乙烯制品的凝胶化程度。在硬质聚氯乙烯制品加工中,聚氯乙烯的塑化程度一直是质量控制的关键。悬浮法合成的聚氯乙烯具有多层次的亚微观结构,存在粉末粒子、约1μm初级粒子、~100nm区域粒子、~10nm微区粒子。微区粒子有一定结晶度。由于聚氯乙烯的热稳定性较差,因此在加工熔融过程中,尽管采取了稳定措施,也很难使微区的晶粒完全熔融。
不同的加工条件往往导致不同的熔融塑化程度。另外,在冷却过程中,已经熔融的微晶又重新结晶,形成与原始晶态不同的结晶度和分布结构。这些微晶可能同时含有几根分子链,形成一种网络结构,使材料具有一定凝胶度。因此聚氯乙烯的熔融塑化过程又称凝胶化过程,由此影响着制品最终的物理力学性能的优劣。长期以来,无法定量测量聚氯乙烯的凝胶化程度,给加工带来很大盲目性。
近年来,在不同温度和不同配方体系下,用实验方法测量聚氯乙烯的凝胶化程度,主要测试实验方法有示差扫描量热法(DSC)和零长毛细管流变仪法。
这里仅介绍零长毛细管流变仪法。零长毛细管流变仪的结构完全同普通毛细管流变仪,只是其配用的毛细管的长径比很小,一般为L/D=0.4。这使得物料通过零长毛细管的流动相当于只是通过毛细管入口区的流动,其压力降几乎全部消耗在入口压力降上。从前面的分析得知,入口压力降的大小主要反映了物料流经入口区时储存弹性变形能的大小,反映了物料弹性的高低。由此可判断,若凝胶化程度高的熔体,其弹性性能好,入口压力降就大;反之,若凝胶化程度低的熔体,其弹性性能差,大入口压力降就小。因此,通过测量聚氯乙烯熔体流经零长毛细管的入口压力降来表征其凝胶化程度,从而进一步表征物料的熔融塑化程度。
用恒压式零长毛细管流变仪,在测试温度160℃和负荷120kg时,测量一种硬聚氯乙烯样品PVC/ACR,图8.2.9给出不同辊温下塑炼的凝胶化程度的数据。由于采用恒压式流变仪,入口压力降效应反映为在恒定压力下熔体通过毛细管的流率大小。熔体弹性高,入口压力损失大,其流率则小,反之则大。图中流率曲线有一个极大值和一个极小值。在低温下塑炼,物料塑化不好,熔体弹性小,流率很大。辊温升高,物料塑化程度变好,熔体弹性变大,流率变小。
若定义流率最大时物料的凝胶化程度为零,而流率最小时凝胶化程度为100%。由下式计算曲线上任一点的凝胶化程度G
图8.2.7给出由式(8.2.19)计算得到的熔体凝胶化程度G。由此图可知,加工温度为140℃时,凝胶化程度等于零,物料塑化很差;180℃时,熔体凝胶化程度相当高。
需要说明的是,这种测量物料凝胶化程度的方法是一种相对测量法。在测量时,由于加热原先塑化好的物料内部结晶结构又会发生变化。由此可见,测量结果与测试温度和预置的恒定压力有关。尽管如此,由于该方法提供了定量比较熔体凝胶化程度的简便途径。因此对于选择硬聚氯乙烯配方体系和工艺条件,尤其是选择硬聚氯乙烯加工性能改性剂,有很大的指导作用。
图8.2.7 PVC/ACR共混物的流率和凝胶度随加工温度的变化[7]
根据入口压力降的特性,巧妙地设计了一种新型双筒毛细管流变仪(Double capillary rheometer),例如英国的Rosand仪器公司。双筒毛细管流变仪有两个料筒,如图8.2.8所示。一个料筒装有大长径比LL/D的普通毛细管。另一个装有零长毛细管,其长径比很少,为LS/D=0.4。两根毛细管的入口区形状相同,两柱塞同时以等速下压挤出熔体。两料筒下端各安装一个压力传感器,测得的压力分别为pL和pS,可以确定毛细管入口压力降为
双毛细管流变仪有两个突出的优点。一方面由于有零长毛细管的对比,使得用普通毛细管测量时的入口压力修正变得十分方便,无须做Bagley修正;另一方面用普通毛细管可以测量熔体的黏度,用零长毛细管可以对比熔体的弹性性能。一次测量同时获得关于熔体黏度、弹性两方面的信息。由入口压力降Δpent可求出熔体入口区的拉伸应力和拉伸黏度等。这里不详细介绍,关于双管毛细管流变仪的测量原理,有兴趣的读者可以参看相关专业书籍和产品说明书。
图8.2.8 双管毛细管流变仪
(a)流变仪(b)结构示意图
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。