



动物源食品中兽药残留是指动物产品的任何可食部分所含兽药的母体、化合物及其降解物。另外,药物或其降解产物与内源大分子共价结合产物称为结合残留,动物组织中存在结合残留则表明药物对靶动物具有潜在毒性作用。抗生素类药物残留是兽药残留的主要代表。动物性食品中的抗生素残留对人体健康的影响,主要表现为过敏毒性作用、细菌耐药性、致畸、致突变和致癌作用等多个方面。由于篇幅有限,本节主要介绍动物源食品中抗生素残留的测定。目前食品中抗生素残留常用的检测方法主要有高效液相色谱法和酶联免疫吸附法。
磺胺喹 啉属磺胺类抗生素兼有抗球虫作用,磺胺喹
啉属磺胺类抗生素兼有抗球虫作用,磺胺喹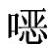 啉纯品为黄色粉末,几乎无味,不溶于水,溶于甲醇、乙醇,易溶于碱性溶液。广泛用于养禽业,其口服后吸收迅速,但排泄缓慢,残留在组织器官及鸡蛋中时间长。近年来我国鸡蛋产量大幅增加,如果蛋鸡场在使用磺胺类抗生素时没有遵守休药期规定,就会造成鸡蛋中药物残留超标,而长期食用有磺胺类抗生素残留的食品对人类健康有害。现介绍农业部1025号公告-15-2008高效液相色谱法检测鸡蛋中磺胺喹
啉纯品为黄色粉末,几乎无味,不溶于水,溶于甲醇、乙醇,易溶于碱性溶液。广泛用于养禽业,其口服后吸收迅速,但排泄缓慢,残留在组织器官及鸡蛋中时间长。近年来我国鸡蛋产量大幅增加,如果蛋鸡场在使用磺胺类抗生素时没有遵守休药期规定,就会造成鸡蛋中药物残留超标,而长期食用有磺胺类抗生素残留的食品对人类健康有害。现介绍农业部1025号公告-15-2008高效液相色谱法检测鸡蛋中磺胺喹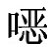 啉残留。
啉残留。
(一)实验原理
供试鸡蛋中残留的磺胺喹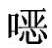 啉经乙酸乙酯提取,过无水硫酸钠柱净化,可用高效液相色谱—紫外法测定,外标法定量。
啉经乙酸乙酯提取,过无水硫酸钠柱净化,可用高效液相色谱—紫外法测定,外标法定量。
(二)试剂与仪器
1.试剂
①所用试剂,除乙腈为色谱纯外,其余均为分析纯试剂。磺胺喹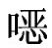 啉含磺胺喹
啉含磺胺喹 啉(C14H12N4O2S)不得少于98.0%。
啉(C14H12N4O2S)不得少于98.0%。
②磺胺喹 啉标准储备液:取磺胺喹
啉标准储备液:取磺胺喹 啉对照品约25mg,精密称定,置250mL量瓶中,用乙腈溶解并稀释成浓度为100μg/mL的储备液。-20℃以下保存,有效期为3个月。
啉对照品约25mg,精密称定,置250mL量瓶中,用乙腈溶解并稀释成浓度为100μg/mL的储备液。-20℃以下保存,有效期为3个月。
③磺胺喹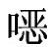 啉标准工作液:精密吸取磺胺喹
啉标准工作液:精密吸取磺胺喹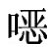 啉标准储备液1.0mL于10mL量瓶中,用流动相稀释成浓度为10μg/mL的标准工作液。
啉标准储备液1.0mL于10mL量瓶中,用流动相稀释成浓度为10μg/mL的标准工作液。
2.仪器与设备
①高效液相色谱仪(配紫外检测器),玻璃层析柱:300mm×10mm,下装G1砂芯板,微孔滤膜:孔径为0.45μm有机系滤膜。
②无水硫酸钠柱的制备:称取无水硫酸钠5g,置入玻璃层析柱中,用约10mL乙酸乙酯淋洗后备用。
(三)操作步骤
1.试料的制备
取适量新鲜的供试鸡蛋内容物,匀浆使均匀,作为供试试料。
取一份适量新鲜的空白鸡蛋内容物作为空白试料。另一份添加适宜浓度的标准工作液,作为空白添加试料。
2.抗生素残留提取
称取试料(5±0.05)g置50mL离心管中,加入20mL乙酸乙酯,匀浆20s,振摇5min,4000r/min离心10min,取上清液过无水硫酸钠柱,收集流出液于鸡心瓶中,残渣再加乙酸乙酯20mL,重复提取一遍,并用少量乙酸乙酯洗残渣,过无水硫酸钠柱,合并流出液于同一鸡心瓶中,于45℃旋转蒸发至近干。残余物用0.015mol/L磷酸溶液-乙腈溶液(1+1)1.0mL溶解,加入1mL水饱和正己烷,涡旋30s,将溶液转移至2.5mL离心管中,5000r/min离心10min,取下层清液,用0.45μm微孔滤膜过滤,供高效液相色谱分析。
3.标准曲线的制备
精密吸取2.0、1.0、0.5、0.2、0.1、0.05、0.025mL磺胺喹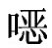 啉标准工作液分别于10mL量瓶中,用流动相稀释成2、1、0.5、0.2、0.1、0.05、0.025μg/mL的浓度,供高效液相色谱分析。
啉标准工作液分别于10mL量瓶中,用流动相稀释成2、1、0.5、0.2、0.1、0.05、0.025μg/mL的浓度,供高效液相色谱分析。
4.测定
(1)色谱条件 C18柱,150mm×4.6mm(i. d.),粒径5μm,或相当者。流动相:0.015mol/L磷酸溶液-乙腈(70+30)。柱温:室温。流速:1.0mL/min。检测波长:270nm。进样量:20μL。
(2)测定方法 取适量试样溶液和相应的标准工作液,做单点或多点校正,以色谱峰面积积分值定量。标准工作液及试样液中磺胺喹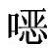 啉的响应值均应在仪器检测的线性范围之内。在上述色谱条件下,磺胺喹
啉的响应值均应在仪器检测的线性范围之内。在上述色谱条件下,磺胺喹 啉的保留时间在5min左右。空白试验:除不加试料外,采用完全相同的测定步骤进行平行操作。
啉的保留时间在5min左右。空白试验:除不加试料外,采用完全相同的测定步骤进行平行操作。
(3)结果计算 按下式计算试料中磺胺喹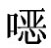 啉的残留量:
啉的残留量:
式中 X——试样中磺胺喹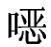 啉的残留量,μg/kg;
啉的残留量,μg/kg;
A——试样溶液中磺胺喹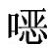 啉的峰面积;
啉的峰面积;
ρs——对照溶液中磺胺喹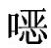 啉的浓度,ng/mL;
啉的浓度,ng/mL;
As——对照溶液中磺胺喹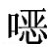 啉的峰面积;
啉的峰面积;
V——溶解残余物的体积,mL;
m——试样的质量,g。
计算结果需扣除空白值,测定结果用三次平行测定的算术平均值表示,保留至小数点后两位。
四环素类药物是以四并苯为母核的一族抗生素,在畜牧业生产中四环素、土霉素、金霉素等主要用于疾病的治疗和预防及促进生长。但该类药物过量使用会导致畜产品中高浓度药物残留,不仅直接影响消费者身体健康,而且会导致细菌耐药性增加。
目前报道的土霉素、四环素等药物残留量的分析方法有微生物法、薄层色谱法、高效液相色谱法、高效液相色谱-串联质谱法和ELISA法等。每种方法各有优缺点,其中ELISA法灵敏度较高、特异性强、测定方法简单快速,可同时筛选检测大量样品。因此,目前常将ELISA法作为筛选方法,高效液相色谱作为准确定量的方法。该检测方法可以同时测定4种兽用四环素类药物残留量。现以乳制品为对象,用高效液相法同时测定其土霉素、四环素、金霉素和强力霉素残留情况,这也是GB/T 22990-2008介绍的方法。
(一)测定原理
用0.1mol/L Na2EDTA-Mcllvaine缓冲溶液提取试样中四环素族抗生素残留,Oasis HLB或相当的固相萃取柱和羧酸型阳离子交换柱,用配有紫外检测器的液相色谱仪测定,外标法定量。
(二)主要试剂和设备
1.试剂
甲醇(色谱纯)、乙腈(色谱纯)、磷酸氢二钠、柠檬酸、乙二胺四乙酸二钠、草酸,所有试剂均为分析纯。
0.2mol/L磷酸氢二钠溶液:称取71.63g磷酸氢二钠,用水溶解,定容至1000mL。
0.1mol/L柠檬酸溶液:称取21.04g柠檬酸,用水溶解,定容至1000mL。
Mcllvaine缓冲溶液:将625mL 0.2mol/L磷酸氢二钠溶液与1000mL 0.1mol/L柠檬酸溶液混合,必要时用NaOH或HCl调pH4.0±0.05。
0.1mol/L Na2EDTA-Mcllvaine缓冲溶液:称取60.50g乙二胺四乙酸二钠放入1625mL Mcllvaine缓冲溶液中,使其溶解,摇匀。
0.01mol/L草酸溶液:称取1.26g草酸,用水溶解,定容至1000mL。
甲醇-水(1+19):量取5mL甲醇与95mL水混匀。
0.01mol/L草酸-乙腈溶液(1+1):量取50mL草酸溶液与50mL乙腈混匀。
0.1mg/mL土霉素、四环素、金霉素、强力霉素(这四个标样纯度均要大于等于96%)标准储备液:准确称取适量四个标准品,分别用甲醇配成0.1mg/mL的标准储备液。储备液于-20℃保存。
土霉素、四环素、金霉素、强力霉素混合标准工作液:根据需要用流动相将土霉素、四环素、金霉素、强力霉素标准储备液稀释成所需浓度的混合标准工作溶液,贮存于冰箱中,每周配制。
Oasis HLB固相萃取柱(或同类柱):500mg,6mL。使用前分别用5mL甲醇和10mL水预处理,保持柱体湿润。
羧酸型阳离子交换柱:500mg,6mL。使用前用5mL甲醇处理,保持柱体湿润。
2.主要设备
液相色谱仪(配有紫外检测器),冷冻离心机(转速大于5000r/min),涡旋振荡器,固相萃取装置,氮气吹干仪等。
(三)测定步骤
1.试样的制备(https://www.xing528.com)
将牛乳从冰箱中取出,放置至室温,摇匀,备用。牛乳置于0~4℃冰箱中避光保存,乳粉常温避光保存。
2.提取
牛乳试样称取10g(精确到0.01g),置于50mL具塞塑料离心管中。乳粉试样称取2g(精确到0.01g),置于50mL具塞塑料离心管中。向试样中加入20mL 0.1mol/L Na2EDTA-Mcllvaine缓冲溶液,于涡旋振荡器上混合2min,于10℃,5000r/min离心10min,上清液过滤至另一离心管中。残渣中再加入20mL缓冲溶液,重复提取一次,合并上清液。
3.净化
将上清液通过处理好的Oasis HLB柱,待上清液完全流出后,用5mL甲醇-水淋洗,弃去全部流出液。减压抽干5min,最后用5mL甲醇洗脱,收集洗脱液于10mL样品管中。
将收集的洗脱液通过羧酸型阳离子交换柱,待洗脱液全部流出后,用5mL甲醇洗柱,减压抽干,用4mL 0.01mol/L草酸-乙腈溶液洗脱,收集洗脱液于10mL样品管中,45℃氮气吹至1.5mL左右,流动相定容至2mL,供液相色谱-紫外检测器测定。
4.测定条件
液相色谱参考条件:色谱柱:Kromasil 100-5C18,5μm,150mm×4.6mm(内径)或相当者;流动相:0.01mol/L草酸溶液-乙腈-甲醇(77+18+5);流速:1.0mL/min;柱温:40℃;检测波长:350nm;进样量:60μL。
5.液相色谱测定
将混合标准工作液分别进样,以浓度为横坐标,峰面积为纵坐标,绘制标准工作曲线,用标准工作曲线对样品进行定量,样品溶液中土霉素、四环素、金霉素、强力霉素的响应值均应在仪器测定的线性范围内。在上述色谱条件下,土霉素、四环素、金霉素、强力霉素的参考保留时间分别为3.09、3.73、8.27和12.53min。
6.结果计算
被测物残留量的测定按下式计算:
式中 Xi——试样中被测组分残留量,μg/k g;
ρ——从标准工作曲线得到的被测组分溶液浓度,ng/mL;
V——样品溶液定容体积,mL;
m——样品溶液所代表试样的质量,g。
磺胺类药物(sulfonamides,SAs)是一类用于预防和治疗细菌感染性疾病的化学治疗药物,它与抗菌增效剂联合使用后,抗菌谱扩大、抗菌活性大大增强,可以从抑菌作用变为杀菌作用。因此,磺胺类药物被广泛应用于兽药临床、动物饲料添加剂、水产养殖等领域。但长期使用不仅会使病菌产生抗药性,而且食品中残留超标易对人类健康造成危害。目前,磺胺类药物残留的检测方法主要有微生物学法、免疫分析法和理化分析法等。GB 29694-2013规定的是高效液相色谱法,它适用于动物源食品中的磺胺醋酰、磺胺吡啶、磺胺甲基吡啶等多残留的检测。
(一)测定原理
样品中残留的磺胺类药物,经乙酸乙醋提取、正己烷脱脂、MCX柱净化后,用高效液相色谱-紫外检测法测定,外标法定量。
(二)主要试剂和设备
1.试剂
乙酸乙酯、乙腈、甲醇、甲酸为色谱纯,其他试剂均为分析纯试剂。磺胺醋酰、磺胺吡啶、磺胺甲基吡啶等标准品含量应≥99%。
MCX柱:60mg/3mL,或相当者。
0.1%甲酸溶液:取甲酸1.0mL,用水溶解并稀释至1000mL。
0.1%甲酸乙腈溶液:取0.1%甲酸830mL,用乙腈溶解并稀释至1000mL。
洗脱液:取氨水5.0mL,用甲醇溶解并稀释至100.0mL。
50%甲醇乙腈溶液:取甲醇50.0mL,用乙腈溶液稀释至100.0mL。
100.0μg/mL磺胺类药物混合标准储备液:取磺胺类药物标准品各10.0mg,于100mL容量瓶中,用乙腈溶解并稀释至刻度。-20℃以下保存,有效期6个月。
10.0μg/mL磺胺类药物混合标准工作液:取100μg/mL磺胺类药物混合标准储备液5.0mL,于50mL容量瓶中,用乙腈稀释至刻度。-20℃以下保存,有效期6个月。
2.仪器和设备
高效液相色谱仪,配紫外检测器或二极管阵列检测器。
旋转蒸发仪、氮吹仪、固相萃取装置。
(三)测定步骤
1.试料的制备
取适量新鲜或解冻待测样品,绞碎后均质。-20℃以下保存待测。取一空白样品,均质后添加适宜浓度的标准工作液,作为空白对照。
2.测定步骤
(1)提取 称取待测试料5.000g,于50mL聚四氟乙烯离心管中,加乙酸乙酯20.0mL,涡动2.0min,4000r/min离心5.0min后将上清液移至100mL鸡心瓶中,残渣中加乙酸乙酯20.0mL,重复提取一次,合并提取液。
(2)净化 在鸡心瓶中加0.1mol/L盐酸溶液4.0mL,于40℃下旋转蒸发浓缩至体积少于3mL,转至10mL的离心管中。用0.1mol/L盐酸溶液2.0mL洗鸡心瓶,转至同一离心管中。再用正己烷3.0mL洗鸡心瓶,将正己烷转至同一离心管中,涡旋混合30s后,3000r/min离心5min,弃正己烷。再次用正己烷3.0mL洗鸡心瓶,转到同一离心管中,涡旋混合30s后,3000r/min离心5min,弃正己烷,取下层液备用。
MCX柱依次用甲醇2.0mL和0.1mol/L盐酸溶液2.0mL活化,取备用液过柱,控制流速1mL/min。依次用0.1mol/L盐酸溶液1.0mL和50%甲醇乙腈溶液2.0mL淋洗,用洗脱液4.0mL洗脱,收集洗脱液,于40℃氮气吹干,加0.1%甲酸乙腈溶液1.0mL溶解残余物,滤膜过滤,供高效液相色谱测定。
3.标准曲线制备
精密量取10μg/mL磺胺类药物混合标准工作液用0.1%甲酸乙腈溶液稀释,分别配制成浓度为10、50、100、250、500、2500、5000μg/L的系列混合标准溶液。以测得的峰面积为纵坐标,对应的标准溶液浓度为横坐标,绘制标准曲线。求回归方程和相关系数。
4.测定
色谱柱:ODS-3C18(250mm×4.5mm,粒径5μm),或相当者;流动相:0.1%甲酸+乙腈,梯度洗脱如表4-14所示;流速:1mL/min;柱温:30℃;检测波长:270nm;进样体积:100μL。
取待测样品溶液和相应的空白对照,作单点或多点校准,按外标法,以峰面积计算。
表4-14 梯度洗脱
5.结果计算
被测物残留量的测定按下式计算:
式中 X——样品中相应的磺胺类药物的残留量,μg/kg;
ρ——样品中相应的磺胺类药物浓度,μg/mL;
V——溶解残余物所用0.1%甲酸乙腈溶液体积,mL;
m——样品质量,g。
计算结果需扣除空白值,测定结果用平行测定后的算术平均值表示,保留三位有效数字。本方法对动物源食品中磺胺类药物残留量的检测限和定量限,肌肉分别为5μg/kg和10μg/kg,内脏分别为12μg/kg和25μg/kg。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。