



棉酚是锦葵科植物棉花的根、茎和种子所含的一种黄色多元酚类有毒化合物。棉酚在棉花中的存在形式可分为游离型和结合型,两者之和即为总棉酚。棉酚在棉籽中含量为0.15%~1.8%,其中棉壳中含0.005%~0.01%,棉仁中含0.5%~2.5%。一般产生有害性的是游离棉酚,所以世界卫生组织(WHO)、联合国粮农组织(FAO)、联合国儿童基金会(UNICEF)规定供人食用的棉籽蛋白制品中游离棉酚(FGP)的含量不得超过0.06%,总棉酚(TGP)不得超过1.2%。如人长期食用含有超标棉酚的棉籽蛋白食品,会产生一系列蓄积性中毒反应。中毒症状为皮肤和胃灼烧、恶心、呕吐、腹泻、头痛,危急时下肢麻痹、昏迷、抽搐、便血,乃至因呼吸、循环系统衰竭而死亡等。
食品及饲料中棉酚的测定方法主要有可见分光光度法、紫外分光光度法及高压液相色谱法等。本实验介绍GB/T 17334-1998用HPLC测定食品中游离棉酚的方法。
(一)检测原理
将食品中的游离棉酚用有机溶液提取后,溶于无水乙醇中,用C18柱将棉酚与杂质分开,在235nm处测定。根据色谱峰的保留时间定性,外标法定量。
(二)主要试剂及仪器
无水乙醇、无水乙醚、棉酚、磷酸均为分析纯,甲醇为HPLC色谱纯,普通氮气。
Agilent 1000高效液相色谱仪,KD-浓缩仪,离心机,100μL微量注射器,Micropark-C18(250mm,6mm)不锈钢色谱柱。
(三)检测步骤
1.样品中游离棉酚的提取
(1)植物油 取油样1.000g,加入5mL无水乙醇,剧烈振摇2min,静置分层,取上清液过滤,离心。
(2)水溶性样品 取样品10.0mL于离心试管中,加入10mL无水乙醚,振摇2min,静置5min,取上层乙醚层,用氮气吹干,用1.0mL无水乙醇溶解,用0.45μm微孔滤膜过滤。
2.标准曲线制备
(1)棉酚标准储备液及工作液的配制 精密称取0.1000g棉酚纯品,用无水乙醚溶解,并定容至100mL,为棉酚标准储备液(1.0mg/mL)。取棉酚储备液5.0mL于100mL容量瓶中,用无水乙醇定容至100mL,为棉酚工作液(50μg/mL)。
(2)色谱分析条件 柱温40℃;流动相,甲醇∶磷酸水溶液=85∶15;测定波长235nm;流量1.0mL/min;进样10μL。
(3)绘制标准曲线 准确吸取2.00,4.00,6.00,8.00,10.00mL的棉酚工作液于10mL容量瓶中,用无水乙醇稀释至刻度,进样10μL,根据响应值绘制标准曲线。
(4)样品分析 取10μL样品溶液进行HPLC分析,根据保留时间确定游离棉酚,根据峰高从标准曲线上查出游离棉酚的含量。
(5)结果计算
式中 X——样品中棉酚的含量,mg/kg;
ρ——从标准曲线查出的游离棉酚的含量,μg/mL;
m——样品质量,g。
(四)注意事项
(1)对油溶性样品用无水乙醇提取其中的游离棉酚,对水溶性产品则用无水乙醚提取。
(2)棉酚提取液及流动相均用0.45μm或0.2μm的微孔滤膜过滤。
河豚毒素(tetrodotoxin,TTX)是豚毒鱼类中的一种神经毒素,为氨基全氢喹唑啉型化合物,分子式C11H17O8N3,相对分子质量319.27。TTX是一种毒性极强的天然毒素,经腹腔注射对小鼠的LD50为8.7μg/kg,其毒性是氰化钠的1000多倍。TTX除在豚毒鱼类中广泛存在外,在两栖动物、软体动物、棘皮动物及甲壳动物等近百种动物中也广泛存在。TTX作为一种钠离子通道阻断剂具有镇痛和解毒等生理功能。TTX主要分布于河豚鱼的肝脏、卵巢、血液和皮肤中。肌肉一般视为无毒,但如鱼体死后时间较长,内脏和血液中的毒素将会慢慢渗入到肌肉中,引起中毒。河豚毒素的毒力单位一般以鼠单位(MU)表示,即在30min内杀死一只20g左右的雄性ddy小鼠的毒素量,根据每克河豚组织中所含毒素的多少,可将河豚组织的毒力分成四个等级:20000MU以上为“猛毒”或“剧毒”;2000~20000MU之间为“强毒”;200~2000MU之间为“弱毒”;100MU以下为“无毒”。据测定,在产卵期间,红鳍圆豚肝脏的毒力为24×106MU,紫圆豚肝脏的毒力为65×106MU,毒性剧烈。
TTX的检测方法主要有小鼠单位法、荧光分光光度法、高压液相色谱法(HPLC)、酶联免疫吸附法(ELISA)、毛细管电泳法(CE)和液相色谱-荧光检测法等。荧光分光光度法定量测定TTX的原理是,TTX在碱性条件下水解后生成2-氨基-6羟甲基-8-羟基喹唑啉(简称C9碱),该物质在370nm光激发下,在495nm处有最大发射波长,通过检测该物质的荧光强度实现TTX的定量。该方法的检出限为0.34~10.0mg/L。除N,N-二甲酰胺可轻度增强荧光外,其他多种试剂对反应均无影响。ELISA法检出限低,可达到0.01mg/L,但需要制备抗TTX的特异性单克隆抗体(McAb)。本实验介绍液相色谱-荧光检测法(GB/T 23217-2008)定量测定TTX。该方法适用于河豚鱼、织纹螺、虾、牡蛎、花蛤、鱿鱼中河豚毒素的测定与确证。本标准方法的检出限为0.05mg/kg。
(一)检测原理
液相色谱-荧光检测法原理是样品中TTX经酸性甲醇提取浓缩后,过C18固相萃取小柱净化,液相色谱柱后衍生,然后用荧光检测器测定,外标法定量。在定量前,本方法还需液相色谱-串联质谱法确证,请参照GB/T 23217-2008介绍。
(二)主要试剂与仪器
1.试剂
甲醇、乙酸、甲酸为色谱纯,其它试剂为分析纯。乙酸铵缓冲液:称取4.6g乙酸铵和2.02g庚烷磺酸钠,加入约700mL水溶解,以乙酸调节pH为5.0,以水稀释至1.0L。4.0mol/L氢氧化钠溶液:称取160g氢氧化钠,以水溶解并稀释至1.0L。河脉毒素标准物质(tetrodotoxin,分子式C11H17N3O8,CAS 4368-28-9):纯度≥98%。
标准储备液(100mg/L):准确称取河豚毒素10.0mg,用少量水溶解后以甲醇定容至100mL,该标准贮备液置于4℃冰箱中保存。标准工作液:根据需要取适量标准储备液,以0.1%甲酸水溶液+甲醇(9+1,体积比)稀释成适当浓度的标准工作液。标准工作液当天现配。基质标准工作液:以空白基质溶液配制适当浓度的标准工作液。基质标准工作液要当天配制。
2.仪器与设备
C18固相萃取柱(500mg/3mL),用前依次以3mL甲醇、3mL 1%乙酸溶液活化,保持柱体湿润,滤膜(0.2μm)及1mL离心超滤管(截留相对分子质量为3000),旋涡振荡器、超声波发生器、减压浓缩装置、固相萃取装置、真空泵(真空度应达到80kPa)、离心机(转速达4000r/min及13000r/min,配有酶标转子)、冷冻高速离心机(转速达到18000r/min,可制冷4℃)。
液相色谱仪(带有荧光检测器与柱后衍生装置)。
(三)检测步骤
1.样品制备
取出有代表性样品可食部分约500g,切成小块,放入组织捣碎机均质,充分混匀,装入清洁容器内,并标记。
2.样品保存
试样于-18℃以下保存,新鲜或冷冻的组织样品可在2~6℃贮存72h。
3.提取
称取5.00g匀浆样品置于50mL聚丙烯离心管中,加入20mL 1%乙酸的甲醇溶液,旋涡振荡2min,50℃水浴超声提取20min,4000r/min离心5min,取上清液,在残渣中再加入20mL 1%乙酸的甲醇溶液,重复以上步骤,合并上清液,过滤至100mL K-D浓缩瓶中,60℃旋转蒸发浓缩至近干,加入2mL 1%乙酸溶液,振荡洗涤浓缩瓶,转移至10mL聚丙烯离心管中,4℃下于18000r/min离心10min,取上清液待净化。
4.净化
将上清液以约1mL/min的流速过柱,用10mL 1%乙酸溶液洗脱,合并流出液与洗脱液,置于25mLK-D浓缩瓶中,于60℃下减压浓缩至近干,用1%乙酸溶液定容1.0mL,过0.2μm滤膜,供液相色谱分析。
5.空白基质溶液的制备
称取阴性样品5.00g,按上述相同的方法操作。
6.测定条件
(1)液相色谱参考条件 色谱柱:Purospher Star PR-18e C18柱,5μm,250mm×4.6mm(内径)或相当者;柱温:30℃;流动相:乙腈-乙酸铵缓冲液(1+19);流速:1.0mL/min;激发波长:385nm,发射波长:505nm;进样量:40.0/μL。
(2)柱后衍生参考条件 衍生溶液:4.0mol/L氢氧化钠溶液;衍生溶液流速:0.5mL/min;衍生管温度:110℃。
(3)色谱测定 根据样品中被测物的含量情况,选取响应值适宜的标准工作液进行色谱分析。标准工作液和待测样液中河豚毒素的响应值应在仪器线性响应范围内。标准工作液与待测样液等体积进样。在上述色谱条件下,河豚毒素的参考保留时间约为10.3min,根据标准溶液色谱峰的保留时间和峰面积,对样液的色谱峰进行定性并外标法定量。
7.确证
由于影响河豚毒素定性及定量的因素较多,在定量前,有条件的实验室还需液相色谱-串联质谱法确证,具体请参照GB/T 23217-2008介绍。
(四)注意事项
样品操作过程中应防止样品受到污染或发生残留物含量的变化。由于河豚毒素为剧毒物质,对于可能含有河豚毒素的产品,应避免直接接触或误食,相关的器皿和器具可以采用4%碳酸钠溶液浸泡加热去毒处理。实验完成后,剩余的实验材料要妥善处理,以免造成中毒。
麻痹性贝类毒素(paralytic shellfish poison,PSP)广泛分布于全球各大海域,是一类对人类健康有较大危害的海洋生物毒素。水体中的藻类是麻痹性贝毒的主要来源,现已知海洋中有13种单细胞的甲藻类可产生麻痹性贝毒。贝类因滤食有毒藻而在体内积累素毒。PSP除存在于贻贝、石房蛤外,还见于某些扇贝、腹足类和蟹类中。PSP在水产动物体内并不是均匀分布的,而是因种类不同、器官不同而有所差异,并有明显的季节变化。对大西洋沿岸的调查表明,软体动物大多数种类的消化系统夏季含毒量最高,鳃和卵巢次之,肌肉含量较低,秋季毒含量下降。
PSP是一类四氢嘌呤的衍生物,到目前为止,已经证实结构的PSP有20多种。根据基团的相似性,PSP可以分为:氨甲酰基类毒素(Carbamoyl compounds):如石房蛤毒素(saxiltoxins,STX)、新石房蛤毒素(neosaxitoxins,neoSTX)、膝沟藻毒素1~4(gonyautoxinsGTX1~4);N-磺酰氨甲酰基类毒素(N-sulfocarbamoyl compounds):如C1~4、GTX5、GTX6;脱氨甲酰基类毒素(decarbamoyl compounds):如dcSTX、dcneoSTX、dcGTX1~4;脱氧脱氨甲酰基类毒素(deoxydecarbomyl compounds):如doSTX、doGTX2,3等。据报道,PSP的毒性因组分不同而差异较大,毒性较高的为氨甲酰基类,而氨甲酰基-N-磺基类毒素的毒性较低。PSP易溶于水且对酸和热稳定,在碱性条件下易分解失活。PSP是一类神经肌肉麻痹剂,其毒理主要是通过对细胞内钠通道的阻断,造成神经系统传输障碍而产生麻痹作用。中毒的临床症状首先是外周麻痹,从嘴唇与四肢的轻微麻刺感和麻木直到肌肉完全丧失力量,呼吸衰竭而死。症状通常在5~30min出现,12h内死亡。PSP的毒性很大,与河豚毒素相当,摄入这种贝毒仅1mg就可导致人死亡。国际许可的安全剂量为100g贝肉中含有相当于80μg以下的STX的毒量。
PSP的检测方法主要有高效液相色谱法、小鼠单位法及细胞毒性试验法等。小鼠生物测试法是检测PSP的传统方法,经过一系列改进后,它是现在唯一国际认可的定量检测PSP方法(Association Official Analytial Chemists,AOAC)。小鼠生物测试法的优点是技术容易掌握、不需要专门的仪器设备,但检测的敏感性与小白鼠的品系关系很大,使得该方法的特异性和精确度不高。利用离子交换型层析法可分离贝类PSP中的STX、neoSTX和GTX1-6,分离的PSP组分在碱性条件下通过氧化作用,可衍生为荧光化合物。将这一氧化反应作为柱后的反应系统与HPLC相结合,即可敏感、专一地定量检测各种PSP毒素。实验证明,柱后衍生HPLC法与生物测试的结果有很好的相关性,灵敏度更高。而且,与其他的检测方法相比,最大的优点是能从少量的粗样中定量分析PSP的毒性成分。但这种方法需要不同结构的PSP毒素标准品。利用高效液相色谱-质谱(LC-MS)则可以直接对样品中的PSP进行分离、定性和定量。目前主要采用动物法测定PSP含量。本实验主要介绍小鼠单位法的测定,以掌握该方法测定贝类PSP的基本技术。
(一)检测原理
GB/T 5009.213-2008规定了麻痹性贝类毒素的检验方法。该标准采用传统的小鼠生物法。鼠单位的定义为:对体重为20g的ICR雄性小鼠腹腔注射1mL麻痹性贝类毒素提取液,使其在15min时杀死小鼠所需的最低毒素量。采用石房蛤毒素(saxitoxin)作为毒素的标准品,将鼠单位换算成毒素的微克数。根据小鼠注射贝类提取液后的死亡时间,查出鼠单位,并按小鼠体重,校正鼠单位,计算确定每100g贝肉内的PSP的微克数。所测的结果代表存在于贝肉内各种化学结构的PSP毒素的含量。
(二)主要试剂及仪器
盐酸、氢氧化钠等均为分析纯。体重19~21g ICR品系的雄性健康小白鼠。
石房蛤毒素(saxitoxin,C10H11N7O4·2HCl)标准溶液(100μg/mL):用蒸馏水水配制20%(体积分数)的乙醇溶液,用5mol/L盐酸调节pH至2.0~4.0,用上述溶液配制石房蛤毒素。
(三)检测步骤
1.样品中PSP的提取
用清水将牡蛎外表彻底洗净,切断闭壳肌,开壳,用清水淋洗内部去除泥沙及其他外来物,取出贝肉。收集约200g肉于10号筛子中沥水5min,检出碎壳等杂物后匀浆。取100g已均质的贝肉于烧杯中,加100mL 0.18mol/L的HCl溶液,充分搅拌,使pH为2.0~4.0。将混合物加热并煮沸5min。冷却后离心5min(3000r/min),取上清液作为样品液。
2.PSP标准工作液的配制
精密称取10.00mg贝类毒素标准品(saxitoxin)于100mL容量瓶中用盐酸酸化的20%乙醇(pH2~4)定容至100mL。此溶液可无限期保存。
用移液管取1mL标准液于100mL容量瓶中,用盐酸酸化的蒸馏水(pH3)定容至100mL,配成1μg/mL的标准工作液。该溶液在3~4℃能稳定数周。分别用10、15、20、25和30mL水稀释10mL浓度为1μg/mL的标准工作液,稀释液的pH应为2~4。
3.中位数死亡时间的标准液选择
将各种稀释度的标准工作液各1mL腹腔注射数只小鼠,选择中位死亡时间为5~7min的浓度剂量。如某浓度稀释液已达到要求,还需以1mL水的增减量进行补充实验。例如:用25mL水稀释的10mL标准液在5~7min内杀死小鼠,还需进行(24+10)和(26+10)的稀释度试验。
以10个小鼠为一组,用中位数死亡时间在5~7min范围内的二种(最好是三种)稀释度的标准液注射小鼠。记录小鼠腹腔注射完毕至停止呼吸的所需死亡时间。
每只小鼠事先称重,精确到0.5g。死亡时间的最短间隔为5s,即7s校正为5s,8s校正为10s,如表4-1所示。
4.毒素转换系数的计算
根据所选稀释液注射的10只小鼠的死亡时间为5~7min之间的死亡时间,由表4-1和表4-2分别查出鼠单位和重量校正因子,两者相乘求得每毫升稀释液的校正鼠单位。将所选稀释液每毫升实际毒素含量的微克数(以Saxitoxin计)除以CMU值便得到毒素转换系数(CF),即CF=μgSAX. /CMU。
计算每组10只小鼠的平均CF值,再取其组间平均值,并以此作为标准作常规检测。
5.小鼠试验
将小鼠称重并记录重量,注射三只小鼠,每只小鼠注射1mL提取液。注射完毕后用秒表记录死亡时间。若小鼠中位死亡时间小于5min,则要进行稀释再注射另一组小鼠。
6.样品中PSP毒力计算
根据每只小鼠的死亡时间,查表4-1得出相应的每毫升注射液的鼠单位数MU,再查表4-2得出质量校正系数。质量校正系数乘以该只小鼠的MU便得到CMU。计算该组小鼠的中位数CMU(将存活鼠的死亡时间视为大于60min)。以中位数按下式计算样品中PSP的含量。
式中 中位数CMU/mL——样品的校正鼠单位;
CF——由标准PSP工作液测定的毒素转换系数;
DF——样品溶液的稀释倍数。
(四)注意事项
(1)S TX作为剧毒品已被收入《化学武器公约》中禁止化学品的第二类清单。S TX标准品及检测到有毒的水产品应妥善处理。
(2)给小鼠做腹腔注射时应熟练操作。若有一滴以上提取液溢出就必须重新注射一只小鼠,并妥善处理所用小鼠。
表4-1 PSP死亡时间—鼠单位的关系
表4-2 小鼠体重校正表
腹泻性贝类毒素(Diarrhetic shellfish poison,DSP)是一类脂溶性物质,其化学结构是聚醚或大环内酯化合物。根据这些成分的碳骨架结构,可以将它们分成三组:酸性成分的大田软海绵酸及其衍生物如大田软海绵酸(okadaic acid,OA)和鳍藻毒素1-3(dinophysistoxin,DTX1-3);中性成分的大环内酯如蛤毒素1-7(pectonotoxin,PTX1-7);磺化毒素成分如扇贝毒素(yessotoxin,YTX)。积累这类毒素的贝类主要有日本栉孔扇贝(Dhlamys nipponensis)、凹线蛤蜊(Mucta sulcatara)、牡蛎(Ostreasp.)、凤螺(StrombusSp)、紫贻贝(Mytilusedulis)、扇贝(Pecten albicans)、中国蛤蜊(Mactrachinensis)及蛤仔(Ruditaoes phlippinarum)等。DSP的直接生产者也是海洋藻类。能产生DSP的藻类主要是甲藻如鳍藻属和原甲藻属的一些藻类。三类毒素的毒理作用各不相同。OA对小鼠腹腔注射的半致死剂量为160μg/g,会使小鼠或其他动物发生腹泻,并且具有强烈的致癌作用。PTX对小鼠的半致死剂量为16~77μg/g,主要作用是肝损伤。磺化毒素对小鼠的半致死剂量是10μg/g,主要破坏动物的心肌。
能引起动物腹泻的DSP主要是OA和DTX1-3。中毒症状为腹泻、呕吐、腹痛和头痛。发病时间在食后0.5~14h不等,一般48h内恢复健康。一般止泻药不能医治。
检测贝类组织中DSP的方法主要是小鼠单位法,即测定小鼠急性中毒的毒量。该方法是AOAC标准方法,也是目前国家规定的标准检测方法(GB/T 5009.212-2008)。其测量单位是鼠单位(MU),即腹腔注射0.5~1.0mL毒素溶液,在注射后使体重16~20g小鼠致死的毒素量,定义为一个鼠单位。1MU相当于3.2μgDTX1。这种方法不需要大型仪器,操作简单,但测定的检出限依赖于小鼠的品系,使结果的重现性差。细胞毒性试验法是基于DSP对某些细胞的毒性,能抑制其生长,毒素剂量与细胞的生长成反比。通过测定细胞的生长来测定毒素的剂量。这种方法与小鼠生物法有极好的相关性,但灵敏度较小鼠生物法差。常用的细胞有L1210小鼠白血病细胞及BGM细胞等。HPLC法是目前用于DSP不同结构组成分析及鉴定最常用的方法。利用HPLC能直接分离贝类原料中的不同结构的DSP,通过将DSP转化成荧光酯类衍生物,通过测定分离组分的荧光强度,并与DSP标准品的荧光衍生物相比较,可实现DSP的定性与定量分析。能与DSP反应生成荧光产物的化合物主要有9-蒽基叠氮甲烷(ADAM)、4-溴甲基-6,7-二甲氧基香豆素(BrDMC)及9-氯甲基蒽(CA)等。由于ADAM极不稳定,而且价格昂贵,BrDMC的选择性不如ADAM,所以现在大多用CA作为衍生化试剂。HPLC结合质谱分析(LC-ISMS),则可直接对贝类中DSP的种类和含量进行分析鉴定。
小鼠单位法在贝类中麻痹性毒素检测时已介绍,本次介绍高效液相色谱分离荧光检测器检测分析贝类中腹泻性贝类毒素含量。
(一)检测原理
贝类组织中的DSP经甲醇提取并用CA衍生化,变成能产生荧光的衍生物,用固相萃取柱提取其中的DSP衍生物后,用反相色谱柱分离,并与标准品比较可确定贝类DSP的种类和含量。
(二)检测材料、试剂及仪器
1.检测材料
扇贝。
2.试剂
OA、DTX1标准品。
甲醇、二氯甲烷、正己烷、无水硫酸钠、9-氯甲基蒽(CA)、四甲基氢氧化胺(TMAH)、乙腈。
3.仪器与设备
电动匀浆器;Beckman 114M高效液相色谱仪[Jasco 821-FP荧光检测器、Beckman210手动进样器、Supelco反相C18柱(4.6mm×150mm,5μm)];硅胶基固相萃取柱(SPE,Supelco,USA)。
(三)检测步骤
1.DSP的提取
将购自市场的鲜活扇贝,用刀切开闭壳肌取出贝肉,称取100g贝肉仔细切取全部中肠腺,将中肠腺匀浆。取1g匀浆后的中肠腺加6mL 80%(体积分数)甲醇后匀浆2min,离心取上清液。残渣加2mL 80%甲醇继续匀浆2min,离心取上清液。合并两次离心上清液。将上清液转入分液漏斗中用15%(体积分数)二氯甲烷/正己烷溶液脱脂三次(每次15mL),加5mL水,再用50%(体积分数)二氯甲烷/正己烷溶液萃取三次(每次15mL)。合并二氯甲烷层,用无水硫酸钠脱水后蒸发至干。用甲醇定容至1mL。得到DSP提取液。
2.DSP的衍生化
取25~50μL样品DSP溶液于1mL Eppendorf管中,于室温下用氮气吹干。加入400μL TMAH溶液(0.8mmol/L,用乙腈稀释),于40℃保温2min,使DSP充分溶解。用氮气吹干。加入400μL CA溶液(0.8mmol/L,用乙腈溶解)于90℃反应1h。冷却至室温,用氮气吹干。加入300μL 50%(体积分数)二氯甲烷/正己烷溶液,溶解衍生化的DSP。
OA和DTX1标准的衍生化按上述同样进行衍生化。
3.衍生化DSP的提取
取两支硅胶基固相萃取柱,一支依次用6mL二氯甲烷和6mL 50%(体积分数)二氯甲烷/正己烷溶液活化(记为A),另一支只用6mL二氯甲烷活化(记为B)。将300μL衍生化的DSP全部转移至A萃取柱中,同时将Eppendorf管用300μL 50%二氯甲烷/正己烷淋洗两次,并将淋洗液全部转移至A萃取柱中,弃去流出液。再分别用6mL 50%二氯甲烷/正己烷溶液和1mL 1%甲醇/二氯甲烷溶液洗A萃取柱一次,并弃去流出液。将B柱与A柱出口相接,在A柱加7mL 5%甲醇/二氯甲烷溶液,收集B柱出口的流出液。室温下用氮气吹干,用2mL流动相溶解,供HPLC分析用。
4.HPLC分析
流动相:乙腈/甲醇(75/25,体积分数),流速1.0mL/min;激发波长:365nm;发射波长:412nm;进样量50μL。
5.DSP组分确定及含量计算
通过与标准DSP洗脱时间对比进行定性,内标法确定含量。
(四)注意事项
(1)进行DSP的衍生化反应之前,加入TMAH的作用是TMAH与DSP形成盐,过量的TMAH会影响后续的衍生化反应。因此在加入TMAH与DSP完全反应后,要用氮气彻底除去过量的TMAH。
(2)在用固相萃取柱提纯DSP衍生物时要注意正确的活化方法。
记忆缺失性贝类毒素(amnesic shellfish toxin,ASP)是一类存在于赤潮藻类中的硅藻属如多列尖刺菱形藻(Nitzsch pungens f.multiseries)中的毒素。中毒症状包括恶心、呕吐、腹痛、腹泻等,同时有昏眩、昏迷等类似神经性中毒症状,永久性丧失部分记忆是此类中毒后的典型特征,因此它被称为记忆缺失性贝类毒素。ASP的主要成分是软骨藻酸(domoicacid,简称DA)是一种具有生理活性的氨基酸类物质。因最早从红藻属的树枝软骨藻(Chondria armata)中分离出来而被命名为软骨藻酸。双壳贝类因滤食海洋微藻而积累ASP。到目前为止,已从紫贻贝(Myltilus edulis)、扇贝(Pecten maximus)、文蛤(Callistachione)等贝类体内以及鳀鱼、鲭鱼和石蟹的内脏中检测到了软骨藻酸。美国食品药品管理局(FDA)将它确定为4种主要海洋生物毒素之一,并规定贝肉中软骨藻酸的安全剂量为20μg/g。我国也于2003年9月2日发布了GB/T 5009.198-2003《贝类记忆丧失性贝类毒素软骨藻酸的测定》。
小白鼠生物检测方法首先被应用于记忆缺失性贝类毒素的检测,该方法是AOAC标准方法。但小鼠生物法的最低检出限为50μg/g,远高于美国FDA规定的贝类食品中ASP的限量标准。为了提高贝类样品中ASP检测的灵敏度,陆续开发了以仪器分析为基础的ASP检测技术。目前,用于贝类食品中ASP检测的仪器分析法主要有高效液相色谱法(HPLC)和毛细管电泳法(CE)。用一定的溶液提取贝类食品中的ASP,用C18反相高效液相色谱柱分离其中的DA。根据DA的紫外吸收特性,用UV检测器进行检测,并通过与标准DA的色谱行为进行比较完成贝类食品中ASP的定性与定量。这种方法的检出限为0.5μg/g,是小鼠生物法的100倍。将贝类的ASP提取液事先用离子交换柱去除部分杂质后,再进行HPLC分析,则检出限可提高到0.02~0.03μg/g。去除杂质后,再用荧光衍生化进行衍生化,则检出限提高到0.006μg/g。毛细管电泳法是近几年发展起来的海洋生物毒素分离检测技术之一,它的基本原理是带电粒子因其不同的质荷比在电场中具有不同的迁移速度而得到分离。软骨藻酸分子中的官能团使其具有质子化的能力,因此它能产生带电粒子并易于毛细管电泳分离。毛细管电泳法的检出限与HPLC法相当。另外,ASP检测还有ELISA检测试剂盒。
本次实验主要介绍贝类食品中ASP的提取、纯化方法和毛细管电泳法测定贝类食品中ASP含量的方法。
(一)检测原理
贝类组织中的ASP(主要是DA)用甲醇溶液提取,并经离子交换树脂纯化,用毛细管电泳分离其中的DA,并与标准DA的电泳行为比较进行定量。
(二)主要试剂与仪器
DA标准品、MUS-1标准物质(贝类组织参照物,DA含量98μg/g)、四硼酸钠(分析纯)、乙腈(色谱纯)、甲醇(色谱纯)、甲酸(色谱纯)。
BioFocus 300自动毛细管电泳仪(Bio-Rad Laboratories),配紫外检测器、组织匀浆器、高速离心机、精密pH计。
阴离子交换柱(SAX:Part No. 1210-2044,Lot No. 182639,Varian)、阳离子交换柱(SCX:Part No. 1211-3039,Lot No. 171069,Varian)、0.45μm微孔滤膜。
(三)检测步骤
1.贝类组织中ASP的提取
将购自市场的鲜活扇贝,用刀切开闭壳肌取出贝肉匀浆,取4g匀浆物加16.0mL甲醇-水(1∶1,体积分数)溶液继续匀浆3min,在4500r/min下离心10min,将上清液用0.45μm微孔滤膜过滤后得到ASP粗提取液。
2.ASP溶液的初步提纯
将SAX柱依次用甲醇、水、甲醇-水(1∶1,体积比)预处理。取5.0mLASP粗提液加入到SAX柱使液体流出,再用5mL乙腈-水(1∶1,体积分数)以1滴/s的速度洗脱杂质。最后,用5mL甲酸溶液(0.1mol/L)洗脱吸附在柱上的DA。
将SCX柱依次用甲醇、水和0.1mol/L甲酸处理。加入5mL从SAX柱上洗脱的DA溶液,用5mL甲酸溶液(0.05mol/L)洗去杂质。最后用6mL的硼酸钠(25mmol/L,pH9.2)-乙腈(9∶1,体积分数)洗脱,收集4~6mL的含DA洗脱液用于毛细管电泳分析。
3.DA的毛细管电泳-紫外检测分析(CE-UV)
非涂渍石英毛细管柱,检测波长242nm,分离电压15.0kV,柱温25℃,压力进样41.37kPa×1s,冲洗及运行缓冲液为25mmol/L硼酸溶液。
在两样电泳条件下,以2μg/mL的DA标准溶液进行CE-UV分析。
4.扇贝样品中DA的定性与定量
通过与标准DA的电泳行为比较进行定性,以外标法进行定量。
硫代葡萄糖苷(glucosinolates,GS)是广泛存在于油菜、甘蓝、芥菜及萝卜等十字花科植物中的一类含硫次级代谢产物。目前已鉴定出的天然硫代葡萄糖苷有100多种。GS在组成上都有一个相同的母体,区别则在于支链R的结构不同。根据R基团的结构特征,可将硫苷分为三大类:脂肪族(第一类)、芳香族(第二类)和吲哚型(第三类)。
GS本身是一稳定的化合物,但在芥子酶(myrosinases)或胃肠道中的细菌酶的催化作用下,会发生降解并生成多种降解产物。硫苷与芥子酶隔离共存于植物体内,当植物的器官受损或对植物加工时它们相接触导致硫苷降解。硫苷和它的降解产物都具有活跃的生物、化学特性。例如,在食品中赋予产品特殊的风味,从而影响食物的适口性。如芥末、辣根的辛辣味,雪菜的雪菜味等。GS的降解产物如5-乙烯基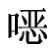 唑硫酮(OZT)及硫氰酸盐等,这些降解产物能抑制甲状腺素的合成和对碘的吸收,从而引起甲状腺肿大。
唑硫酮(OZT)及硫氰酸盐等,这些降解产物能抑制甲状腺素的合成和对碘的吸收,从而引起甲状腺肿大。
GS的测定方法有高效液相色谱(HPLC)法和比色法。HPLC法主要用沸腾的甲醇溶液提取样品中的硫代葡萄糖苷,并将提取的GS溶液用离子交换柱进行预处理,然后用配备紫外检测器的反相高效液相色谱(RP-HPLC)仪测定。在此方法中,选用了硫代葡萄糖苷类化合物中的一种,即带有一个结晶水的黑芥子硫苷酸钾(相对分子质量415.49)作为定量分析的内标物。经过多个国际权威实验室的协同试验,共同确定了16种以上硫代葡萄糖苷类化合物的仪器保留时间,将色谱图上各类硫代葡萄糖苷的峰面积根据内标物峰面积校正,计算得到其单独含量,累加得到硫代葡萄糖苷类化合物的总量,均以μmol/g为单位。该方法已于1992年被确认为国际标准(ISO),目前是加拿大谷物委员会(CGC)谷物研究实验室(GRL)出具加拿大双低油菜籽年度质量分析报告时所使用的基准方法。这种方法的优点是能对蔬菜样品中的各种GS进行定性和定量分析。
高效液相色谱法是目前报道较多的测定方法。如,我国农业部规定的油菜籽中硫代葡萄糖苷含量的方法(NT/T 1582-2007)。本实验介绍高效液相色谱法测定GS总量的方法。
(一)检测原理
用甲醇-水溶液,提取硫代葡萄糖苷,然后在阴离子交换树脂上纯化并酶解脱去硫酸根,再经反相C18柱分离,由紫外检测器检测硫代葡萄糖苷,内标法定量。
(二)试剂及仪器(https://www.xing528.com)
1.试剂及溶液配制
硫酸酯酶(EC3.1.6.1),每毫升硫酸酯酶活性单位不低于0.5,硫酸酯酶溶液应即配即用。葡聚糖凝胶悬浮液:称取10g DEAE SephadexG-25葡聚糖凝胶,浸泡在过量的2mol/L醋酸溶液中,静置沉淀,再加入2mol/L醋酸溶液,直到液体体积是沉淀体积的2倍,于4℃冰箱中存放,待用。甲醇-水溶液(70+30)。0.02mol/L醋酸钠溶液:称取0.272g醋酸钠(CH3COONa·3H2O),加入800mL水溶解,用醋酸调节溶液的pH至4.0,加水定容至1L。6.0mol/L甲酸咪唑溶液:称取204g咪唑,溶解于113mL甲酸中,待溶液冷却后加水定容至500mL。
用丙烯基硫代葡萄糖苷(相对分子质量415.49)作内标,当样品中含有丙烯基硫代葡萄糖苷时,用苯甲基硫代葡萄糖苷(相对分子质量447.52)作内标。当样品硫代葡萄糖苷含量低于20.0pmol/g时,可将丙烯基硫代葡萄糖苷等内标物质浓度降低为1.0~3.0mmol/L。内标溶液在4℃的冰箱中可存放3周,在-18℃条件下可保存更长时间,内标溶液的纯度检定参照表4-3执行。
表4-3 脱硫硫代葡萄糖苷的相对校正系数
5.0mmol/L丙烯基硫代葡萄糖苷内标溶液:称取207.7mg丙烯基硫代葡萄糖苷溶解于80mL水中,加水定容至100mL。20.0mmol/L丙烯基硫代葡萄糖苷溶液:称取831.0mg丙烯基硫代葡萄糖苷溶解于80mL水中,加水定容至100mL。35mmol/L苯甲基硫代葡萄糖苷溶液:准确称取223.7mg苯甲基硫代葡萄糖苷溶解于80mL水中,加水定容至100mL。20mmol/L苯甲基硫代葡萄糖苷溶液:准确称取895.0mg苯甲基硫代葡萄糖苷溶解于80mL水中,加水定容至100mL。
流动相A:超声波脱气30s的水。流动相取200mL色谱级乙腈,加入800mL水,混匀,超声波脱气30s。
2.仪器与设备
聚丙烯离子交换微柱(底部筛板为100目),离心机(5000g),0.45μm水溶性微孔滤膜,色谱柱:填料颗粒小于或等于10的反相C18或Cs柱,高效液相色谱仪(具备梯度洗脱,柱温可控制在30℃,带紫外检测器)。
(三)检测步骤
1.硫代葡萄糖苷的提取
分别称取200.0mg待测试样至A、B两支离心管中。将离心管于75℃水浴1.0min,加入2.0mL沸甲醇-水溶液(70+30)后,立即加入200μL 5mmol/L内标溶液至A管中,200μL 20mmol/L内标溶液至B管中。75℃水浴10min,其间每隔2min取出离心管在旋涡混合器上旋涡混合,然后取出离心管冷却至室温,5000×g离心3min,分别转移上清液至10mL刻度试管A′、B′中。
分别向A、B管中再加入2.0mL70%沸甲醇-水溶液(70+30),75℃水浴约30s,涡旋混匀后,75℃水浴10min,其间每隔2min取出涡旋混合,取出离心管冷却至室温,5000g离心3min,分别转移上清液至原刻度试管A′、B′中。用水调节A′、B′管中的提取液至5.0mL,混匀。此提取液在低于-18℃的暗处可保存2周。
2.离子交换柱的制备
将聚丙烯离子交换柱垂直置于试管架上。取0.5mL充分混匀的葡聚糖凝胶悬浮液至每一离子交换柱中(勿黏附柱壁)。静置待液体排干后,取2.0mL 6mol/L甲酸咪唑溶液冲洗树脂,排干后,再用1.0mL水冲洗树脂两次,每次均让水分排干。
3.纯化、脱硫酸根
取1.0mL提取液缓缓加入已准备好的离子交换微柱中(勿搅动表面),待液体排干后,分别加入1mL 0.02mol/L醋酸钠溶液两次,每次加入后均让液体排干。加入100pL硫酸酯酶溶液至离子交换微柱,35℃条件下反应16h。分别用1.0mL水冲洗离子交换微柱2次,洗脱液收集于试管中。用水将洗脱液定容至5.0mL,充分混匀后,用0.45μm的微孔滤膜过滤,待进样。洗脱液在-18℃暗处可存放1周。
4.空白试验
用相同的样品进行相同的前处理,但不加内标物质,以检定样品中内标物质是否存在。
5.色谱测定
(1)仪器条件 流动相流速为1.0mL/min,柱温30℃,紫外检测器检测波长229nm。
(2)洗脱梯度 不同有分离柱,洗脱梯度不同。Spherisorb C18柱,10μm(150mm×4mm)和Novapak C18柱,5μm(150mm×3.9mm),洗脱梯度见表4-4。LichrosorbRp18柱,5μm(150mm×6mm),洗脱梯度见表4-5。Lichrospher Rp18柱,5μm(125mm×4mm),洗脱梯度见表4-6。
表4-4 Spherisorb C18柱和Novapak C18柱洗脱梯度
表4-5 Lichrosorb Rp18柱洗脱梯度
表4-6 Lichrospher Rp8柱洗脱梯度
(3)色谱测定 进样量10μL,记录峰面积。内标法定量。
6.结果计算
(1)单组分硫代葡萄糖苷含量的计算
①以每克干基脱脂油菜籽中所含硫代葡萄糖苷的物质的量(μmol)表示,按下式计算,计算结果精确到小数点后两位。
式中 D1——干基脱脂油菜籽中硫代葡萄糖苷含量的数值,μmol/g;
Ag——硫代葡萄糖苷峰面积;
As——内标峰面积;
Kg——脱硫硫代葡萄糖苷相对校正系数,如表4-3所示;
m——试样质量,g;
n——试样中加入内标物的量,μmol;
w——试样中水分、挥发物和含油量之和,%。
②以每克脱脂油菜籽含标准水分及挥发物时所含硫代葡萄糖苷的物质的量(μmol)表示,按下式计算,计算结果精确到小数点后两位。
式中 Ag、As、n、m、Kg、w同上式;
D2——脱脂油菜籽含标准水分及挥发物时硫代葡萄糖苷的含量,μmol/g;
Ws——标准水分及挥发物含量,以质量分数表示,数值为8.5%或9%。
③以每克干基油菜籽中所含硫代葡萄糖苷的物质的量(μmol)表示,按下式计算,计算结果精确到小数点后两位。
式中 Ag、As、n、m、Kg同上式;
D3——干基油菜籽中硫代葡萄糖苷含量,μmol/g;
Wt——试样中水分及挥发物含量的数值,%。
④以每克油菜籽含标准水分及挥发物时所含硫代葡萄糖苷的物质的量(μmol)表示,按下式计算,计算结果精确到小数点后两位。
式中 Ag、As、n、m、Kg、Wt、Ws同上式;
D4——油菜籽含标准水分及挥发物时硫代葡萄糖苷的含量的数值,μmol/g。
(2)硫代葡萄糖苷含量的计算 硫代葡萄糖苷含量等于单组分硫代葡萄糖苷(单组分峰面积应大于峰面积总和的1%)含量的总和,以每克样品中所含硫代葡萄糖苷的微摩尔数表示。如果A、B两管硫代葡萄糖苷含量的测定值满足下列精密度的要求,硫代葡萄糖苷的含量为两测定值的算术平均值。计算结果精确到小数点后两位。
精密度要求:
①同一试样、同一方法、同一操作者、同一仪器、同一实验室短期内两次测定值:如果硫代葡萄糖苷含量低于20.00μmol/g,绝对差值不大于2.00μmol/g;如果硫代葡萄糖苷含量在20.00~35.00μmol/g范围内,绝对差值不大于4.00μmol/g;如果硫代葡萄糖苷含量大于35.00μmol/g,绝对差值不大于6.00μmol/g。
②同一试样、同一方法、不同操作者、不同仪器、不同实验室两次测定值:如果硫代葡萄糖苷含量低于20.00μmol/g,绝对差值不大于4.00μmol/g;如果硫代葡萄糖苷含量在20.00~35.00μmol/g范围内,绝对差值不大于8.00μmol/g;如果硫代葡萄糖苷含量大于35.00μmol/g,绝对差值不大于12.00μmol/g。
氰苷是由腈醇(α-羟基腈)上的羟基和D-葡萄糖缩合而成的β-糖苷衍生物。氰苷广泛存在于豆科、蔷薇科、禾本科等的100多种植物中。含有氰苷的食源性植物有木薯、豆类和一些果树的种子如杏仁、桃仁、亚麻仁等。另外,一些鱼类如青鱼、草鱼、鲢鱼等的胆汁中也含有氰苷。常见的氰苷有苦杏仁苷(amygdalin)、亚麻苦苷(linamarin)、高粱苦苷(dhurrin)等。氰苷能通过生氰作用而产生毒性很强的氰氢酸(HCN),从而造成对人体的危害。
(一)检测原理
氰化物在酸性条件下被蒸出吸收于碱性溶液中。在pH7.0的溶液中,用氯胺T将氰化物转变为氯化氢,再与异烟酸-吡唑酮作用生成蓝色,与标准系列进行比较定量。
(二)主要试剂及仪器
试银灵(对二甲氨基亚苄罗丹宁)、丙酮、异烟酸、氢氧化钠、吡唑酮、N-二甲基甲酰胺、氰化钾、乙酸锌、酒石酸、乙酸、氯胺T,均为分析纯。
蒸馏装置、721分光光度计。
(三)检测步骤
1.标准曲线的制作
(1)异烟酸-吡唑酮溶液的配制 取1.5g异烟酸溶于24mL 2%氢氧化钠溶液中,加水至100mL。另取0.25g吡唑酮溶于200mLN-二甲基甲酰胺中,合并上述两种溶液,混匀。
(2)氰化钾标准溶液的配制 取0.25g氰化钾溶于水并定容至3000mL,使用前用0.1%氢氧化钠溶液稀释成1μg/mL氢氰酸,作为氰化钾标准溶液。
(3)标准曲线的绘制 取0、0.3、0.6、0.9、1.2、1.5mL氰化钾标准溶液于25mL比色管中,用水补足至10mL。于上述各比色管中各加1mL 1%氢氧化钠溶液和1滴酚酞指示剂,用1.5%乙酸调至红色消失。加5mL磷酸盐缓冲液(0.5mol/L,pH7.0),置于37℃水浴中,再加入0.25mL氯胺T溶液,混匀后保温5min。加入5mL异烟酸-吡唑酮溶液,加水至25mL,于20~40℃下放置40min。以2cm比色杯,零管调节零点后,于638nm处测吸光度,并绘制标准曲线。
2.样品中氰化物提取
取10.0g样品,置于250mL蒸馏瓶中,加适量水使样品全部浸没。加20mL乙酸锌(0.1g/mL),1~2g酒石酸。迅速连接好蒸馏装置,冷凝管下端插入5mL氢氧化钠溶液(0.1g/mL)中,开始蒸馏。收集馏出液近100mL,并定容至100mL。
3.样品中氰化物的测定
取10mL提取液于25mL比色管中,按标准曲线制作方法进行比色。
4.样品中氰化物的含量计算
式中 A——从标准曲线上查得的氢氰酸的浓度;
m——样品质量,g;
V1——测定用蒸馏液体积;
V2——样品蒸馏液总体积。
(四)注意事项
(1)在氰化物的显色反应中,pH对显色结果影响较大,加入的磷酸盐缓冲液的pH要准确。
(2)氯胺T的有效含氯量对测定结果影响较大,氯胺T溶液应现用现配。
龙葵碱又叫茄碱、龙葵毒素、马铃薯毒素,是由葡萄糖残基和茄啶形成的一种弱碱性糖苷。龙葵碱广泛存在于马铃薯、番茄及茄子等茄科植物中。龙葵碱在马铃薯中的含量一般在0.005%~0.01%之间,马铃薯发芽后,其幼芽和芽眼部分含量可达0.3%~0.5%。龙葵碱对胃肠道黏膜有较强的刺激作用,对中枢神经也有一定的麻醉作用,是马铃薯能引起食物中毒的主要因素。本次介绍比色法测定龙葵碱的方法。
(一)检测原理
龙葵碱不溶于水、乙醚及氯仿,但能溶于乙醇。利用乙醇提取马铃薯中的龙葵碱,提取的龙葵碱在稀硫酸中与甲醛作用生成橙红色化合物,在一定浓度范围内,颜色的深浅与龙葵碱的浓度成正比。
(二)主要试剂及仪器
95%乙醇、冰乙酸、硫酸、甲醛、龙葵碱标准品、浓氨水。
匀浆器、离心机、旋转蒸发器、721分光光度计。
(三)检测方法
1.龙葵碱标准曲线的制作
(1)龙葵碱标准溶液的配制 精确称取0.1000g龙葵碱,以1%硫酸溶液溶解并定容至1000mL,配成100μg/mL的龙葵碱标准溶液。
(2)标准曲线的制作 分别取0、0.1、0.2、0.3、0.4、0.5mL龙葵碱标准溶液于10mL比色管中,用1%硫酸补足至2mL,在冰浴中滴加浓硫酸至5mL(滴加速度要慢,时间应不少于3min),摇匀。静置1min,然后在冰浴中滴加1%甲醛溶液2.5mL。静置90min,于520nm下测吸光度,并绘制标准曲线。
2.样品中龙葵碱的提取
取捣碎的马铃薯样品20g于匀浆器中加100mL95%乙醇,匀浆3min。将匀浆液离心10min(4000r/min),取上清液,残渣用20mL乙醇洗涤二次,合并上清液。将上清液于旋转蒸发器上浓缩至干。用5%硫酸溶解残渣,过滤后将滤液用浓氨水调至中性,再加1~2滴浓氨水使pH至10~10.4,在80℃水浴中加热5min,冷却后置冰箱中过夜,使龙葵碱沉淀完全。离心,倾去上清液,以1%氨水洗至无色透明,将残渣用1%硫酸溶解并定容至10mL。得龙葵碱提取液。
3.样品龙葵碱的测定
取龙葵碱提取液2mL于10mL比色管中。按标准曲线制作方法,测定其吸光度。
4.样品龙葵碱含量计算
式中 m0——从标准曲线上查得的龙葵碱的量,μg;
m——样品质量,g。
生物胺是一类具有生物活性的含氮有机化合物的总称,主要是氨基酸脱羧酶对游离氨基酸脱羧反应的产物。生物胺在食品中广泛分布,常见的有组胺、酪胺、腐胺、尸胺、精胺、亚精胺、色胺和苯乙胺等,其中,水产品中的生物胺含量尤其高。当人体摄入过量生物胺,会导致脸红、呕吐、呼吸加快、支气管痉挛、头痛以及高血压等中毒症状,尸胺、腐胺、精胺和亚精胺等生物胺还会与亚硝酸盐反应生成致癌物亚硝胺。由于生物胺本身无紫外吸收,本实验采用丹磺酰氯对食品中常见的八种生物胺先进行衍生,用高效液相色谱法测定。
(一)检测原理
食品中的生物胺用酸性介质提取后,通过缓冲液调整为碱性体系,使用丹磺酰氯进行柱前衍生,再通过高效液相色谱进行分离和测定,与标准系列比较定量。
(二)主要试剂与仪器
丹磺酰氯、高氯酸、正庚烷、碳酸钠、碳酸氢钠、碳酸钾、色谱级甲醇、超纯水、生物胺标准品。
高效液相色谱仪、氮吹仪、色谱柱。
(三)检测方法
1.试剂的配制
(1)生物胺标准液 使用0.1mol/L盐酸配制终浓度分别为0.25、1、2.5、5、10、15、25和50mg/L的生物胺,4℃冰箱避光保存。
(2)丹磺酰氯衍生剂溶液 以丙酮为溶剂将丹磺酰氯配制成10mg/mL衍生剂溶液,4℃冰箱避光保存。
(3)pH11缓冲液500mL的0.5mol/L碳酸氢钠和120mL的0.5mol/L碳酸钠混合均匀,使用碳酸钠溶液调节pH为9.2,置于冰箱中可以保存数月。使用前,每50mL上述缓冲液中加入16.65g碳酸钾,缓冲液的pH即为11.0~11.1,必须现配现用。
(4)脯氨酸溶液 以超纯水为溶剂将脯氨酸配制成100mg/mL的溶液。
2.生物胺标准曲线的制作
(1)生物胺衍生 取1.00mL生物胺标准液,加入1.50mL pH11的缓冲液,混合均匀,然后再加入1mL丹磺酰氯衍生剂溶液,40℃水浴暗反应1h;加入0.20mL脯氨酸溶液,混合均匀,室温保持1h;加入3.00mL正庚烷萃取,涡旋混合,分层后取1.00mL上层有机相,40℃下氮气吹干;加入1.00mL甲醇溶解残留物,过0.22μm有机膜,待上机测定。
(2)生物胺测定 Capcell Pak C18MGⅡ(4.6mm×150mm,5μm)色谱柱;流速:0.80mL/min;进样量:10μL;柱温:40℃;荧光检测器参数:Ex=350nm,Em=520nm;采用超纯水(A)和甲醇(B)作为流动相,梯度洗脱程序如下:0min 45%A+55%B,7min 35%A+65%B,14min 30%A+70%B,20min 30%A+70%B,27min 10%A+90%B,30min 0%A+100%B,31min 0%A+100%B,32min 45%A+55%B,37min 45%A+55%B。
(3)标准曲线制作 以各生物胺的峰面积为横坐标、浓度为纵坐标绘制标准曲线。
3.样品中生物胺的提取和测定
准确称取1.00g样品,加入4.00mL的0.4mol/L高氯酸,均质,4℃、3500g离心10min,取上清液,再次向沉淀中加入4.00mL的0.4mol/L高氯酸,均质,4℃、3500g离心10min,两次上清液合并后定容至10mL,作为生物胺提取液。取1.00mL提取液按照2中的方法进行衍生和测定。
4.样品中生物胺含量的计算
式中 ρ——从标准曲线查得的生物胺含量,mg/L;
V——样品提取过程中两次上清液合并后定容的体积,mL;
m——样品质量,g。
(四)注意事项
(1)生物胺衍生必须在强碱性的环境下避光进行。
(2)流动相使用的甲醇必须是色谱纯的,水必须是超纯水。
(3)样品上机测试前必须过0.22μm有机膜,防止污染高效液相色谱系统或色谱柱。
食品过敏是由于摄入一种或多种含有过敏原的食物而引起的以免疫损伤为主要表现形式的免疫应答。其主要表现形式为皮肤反应(荨麻疹、血管性水肿、湿疹),呼吸症状(哮喘、鼻炎),肠胃症状(呕吐、腹泻、肠胃痉挛),系统反应(心血管症状包括过敏性休克)等。在过去的几十年中,食品过敏的发病率和流行情况日益增加,食品过敏现已成为一个新兴的公众性健康问题。随着世界食品贸易的发展,人们生活习惯的改变,饮食面的快速拓宽,特别是转基因食品的大量涌现,过敏症状趋于多样化、复杂化和严重化。
据联合国粮农组织报告,在引起人们过敏的食物中,超过90%的食物过敏是由八大类主要的食物引起的,这八大类食物主要包括牛乳及乳制品、鸡蛋及蛋制品、鱼及鱼制品、甲壳类及制品、花生、大豆、坚果、谷物及其制品。目前还没有治疗食物过敏的有效的方法,唯一有效的方式是避免食用和接触能够引起过敏的食物。因此,对食品过敏原的检测与分析就是一个极其重要的问题。
食物过敏原的检测方法主要有酶联免疫法(ELISA)、火箭免疫电泳(rocket immune-electrophoresis,RIEP)、PCR法、组胺释放试验及过敏原指纹图谱快速检测方法等。本实验主要介绍的是酶联免疫的分析方法。
(一)检测原理
食品试样中的过敏原采用磷酸盐缓冲液提取,提取液经离心后取上层清液,将一定量的上清液加入固定有过敏原多克隆抗体的酶标板中,孵育一段时间后,加入HRP标记的过敏原单克隆抗体,然后加入显色底物,用酶标仪进行定量测定。
(二)试剂及仪器
酶标单克隆抗体:HRP-抗过敏原-IgG;过敏原多克隆抗体(兔);待检食品、样品抽提液:0.01mol/L磷酸盐缓冲液。
包被液:pH9.50.05mol/L碳酸盐缓冲液;洗涤液:pH7.40.02mol/L Tris-HCl缓冲液+0.05%吐温;封闭液:pH7.40.02mol/L Tris-HCl缓冲液+0.2%BSA;底物缓冲液:0.1mol/L Na2HPO4(Na2HPO4·12H2O 35.8g/L)5.14mL,0.05mol/L枸橼酸(10.5g/L)4.86mL混匀,加入邻苯二胺(OPD)4mg,置棕色小瓶中,临用时加30% H2O24.0μL,混匀;终止液:2mol/L H2SO4。
恒温振荡培养箱、恒温箱、酶标反应板、酶标分光光度计、组织捣碎机。
(三)检测步骤
1.样品的抽提
将食品在组织捣碎机中捣碎后,称取一定量的糜状样品,加入5∶1(体积/重量)的0.01mol/L磷酸盐缓冲液充分匀浆,在25℃恒温振荡培养箱中抽提2h(120r/min)。抽提液于4℃以9000r/min离心20min,上清液过0.45μm的滤膜,考马斯亮蓝法测定蛋白含量,分装-20℃冻存。
2.酶联免疫步骤的操作
(1)恒温箱取包被液适当稀释的抗过敏原多克隆抗体100μL,加到酶标板的各孔中,加盖,4℃过夜。次日用洗涤液充分洗涤3次,甩干。
(2)各孔内加不同稀释倍数的待检过敏原抽提物100μL,同时加阳性和阴性对照样品,置37℃恒温箱中孵育60min,移去液体。同前法洗涤3次,甩干。
(3)各孔内加稀释HRP-抗过敏原单克隆抗体100μL,置37℃恒温箱中孵育60min。移去液体,同前法洗3次,甩干。
(4)各孔加底物缓冲液100μL,置黑暗处20min。
(5)各孔内加2mol/L H2SO450μL,终止反应。
3.结果判定
肉眼判定:可于白色背景上,直接用肉眼观察结果:反应孔内颜色越深,阳性程度越强,阴性反应为无色或极浅,依据所呈颜色的深浅,以“+”“-”号表示。
酶标仪检测:在ELISA检测仪上于492nm处,以空白对照孔调零后测各孔OD值,若大于规定的阴性对照OD值的2.1倍,即为阳性,否则为阴性。
(四)注意事项
(1)该方法定量测定食物过敏原的检测限为10μg/kg。
(2)显色液的配制及H2O2的加入时间要格外注意。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。