



鞣制机理,是制革工作者长期以来所致力研究的重大课题。研究鞣制机理,旨在揭示鞣制过程的物理化学本质。
迄今为止,不少研究者通过各种实验方法和研究途径从不同的深度和广度,做了大量研究工作,提出许多解释鞣制本质的假说和理论,为现代鞣制理论奠定了基础。
众所周知,鞣制过程的微观世界是相当复杂的。它包含鞣制系统的复相反应与多成分平衡,涉及胶原与鞣剂的结构、性质和鞣制化学热力学与动力学,是一个物理化学总体系。但是,鞣制的核心问题则仍然是鞣剂质点向裸皮内部渗透及其与胶原结构的相互作用。由于胶原与鞣剂本身结构的复杂性,有关鞣制化学热力学和动力学研究的难度较大,加上鞣制反应的许多影响因素,因此目前的鞣制机理研究尚处于间接推证阶段。
本文并非从物理化学的研究深度来论述鞣制机理,而是基于现有研究和有关的资料,就鞣制机理方面的问题,作初步的综合和阐述,以资探讨。
“纯粹的量的分割是有一个极限的,到了这个极限它就转化为质的差别。”(恩格斯:《自然辩证法》)生皮经过鞣前一系列湿加工(即准备工段加工),并没有发生“质的变化”。不仅没有完成由皮到革的转化,相反,胶原结构中原有的结合键却遭到削弱,乃至部分被破坏,导致胶原内聚力减小,收缩温度降低。这种“量的分割”以鞣制为“极限”,一旦鞣制开始,它就随着鞣剂与胶原的相互作用而向“质的变化”方面转化。当鞣剂质点与胶原彼此形成足够的不可逆的交联键时,胶原发生改性,结构获得加固,于是“质的变化”开始到来,鞣制效应也随着产生,生皮便转变成具有物理强度、耐湿热性和抗化学作用性的革。
那么,何谓鞣制效应呢?按照鞣制交联理论,鞣制效应的发生至少必须满足两个条件:(1)鞣剂质点必须与胶原结构中两个或两个以上的反应点作用,并彼此形成交联键;(2)表征鞣制效应的交联作用必须是不可逆的。
基于上述,所谓鞣制效应,其宏观概念就是裸皮转化成为具有物理强度、耐湿热性和抗化学作用性的革;其微观概念就是鞣剂质点与胶原结构中两个以上反应点的不可逆的交联作用,“网络”了胶原结构,增大了胶原的内聚力,致使胶原发生变性。
交联理论认为,当鞣剂质点仅以沉淀物的形态沉积于胶原结构中,抑或仅与胶原结构中一个反应点结合、抑或交联作用具有明显的可逆性时,由于它们没有满足产生鞣制效应的两个必须条件,所以都不能列为鞣制效应。这种非交联作用的沉积与结合,被认为是填充效应。填充效应虽然不足以显示革的耐湿热特性,但是对于增加胶原对鞣剂的吸收量、提高成革的丰满性和弹性、加强鞣制效果是有助益的。
交联理论解释鞣制效应的基本论点,已为大量的实验结果和生产实践所证实。据此,对于鞣制本质的认识,可以认为,鞣制效应取决于鞣剂质点与胶原的多点交联结合;而鞣剂质点在胶原结构内的沉积与非交联结合则在很大程度上加强了鞣制效果。
胶原与鞣剂是鞣制系统的主体。决定鞣制反应及其进程的内在因素,就是胶原与鞣剂的组成、结构和性质,以及它们之间的物理化学作用。
(一)胶原
对胶原的研究表明,胶原是一个具有三根多肽链的三股螺旋体。每根多肽链约由1052个不同的氨基酸组成,其中具有“—RCH—NH—CO—”的重复结构。多肽链又称主链,相邻主链间的中心距离为1.1~1.6nm。每个主链上,约有162个极性氨基酸的侧链。由于胶原的三个主链是末端对齐排列的,因而极性氨基酸的分布也成为均匀而有顺序。胶原侧链上的极性氨基酸残基,决定胶原为两性离子化合物。
就鞣制反应而言,胶原相邻主链间的中心距离,是鞣剂质点藉以进入胶原内部并与之产生交联的必要空间。不难想象,当鞣剂质点的基本长度过大或过小于胶原相邻主链间的中心距离时,那么鞣剂质点与胶原相邻主链的交联结合就成为不可能,而胶原主链上的酰胺基(—NH—CO—)和侧链上的氨基酸残基[主要是羧基(—COOH)、氨基(—NH2)、羟基(—OH)]却是参与鞣制反应的主要官能团。由此可见松散胶原纤维的重要性。
胶原的氨基酸组成及其三重螺旋体结构,决定它是多官能团的化合物。已经确认,在鞣制过程中,参与鞣制反应的胶原官能团如表1所列。
表1仅仅对胶原参与鞣制反应的官能团位置及其大致化学键型作定性说明,至于各种官能团参与鞣制反应的定量关系,尚需根据具体鞣法作深入研究。
表1 参与鞣制反应的胶原官能团
(二)鞣剂
鞣剂是一个范围很广的概念。不同鞣剂,其组分、结构、性质各异。但是,任何鞣剂的结构中必然具有能与胶原相应官能团作用的鞣性基团。鞣制效应主要取决于鞣剂结构中的鞣性基团,但鞣剂的性质及其鞣革质量,却很大程度上决定于鞣剂中包括非鞣性基团、母体等整个化学结构,这个道理为具有一般化学知识者所熟知。
在常见的鞣剂中,大体可划分为无机鞣剂与有机鞣剂两大类。无机鞣剂主要为铬、锆、铝、铁、钛等金属盐类;非金属的硅、磷、硫等化合物,目前在实际生产中应用甚少。有机鞣剂主要为植物鞣剂、醛鞣剂、油鞣剂和合成鞣剂。
根据鞣剂化学的研究,无机鞣剂中的金属盐类都具有络合物的结构和性质(非金属硅也能形成络合物,磷则以含氧酸的多聚体形态显示鞣性);有机鞣剂根据芳族或脂族的体系具有醇、醚、醛、酮、酸、酯的结构和性质。
1.无机鞣剂
以铬络合物为代表。络合物化学表明形成体,以中心离子为Cr3+形成体的单核铬络合物,其配位数为6,铬离子的空间结构为正八面体。随着络合物内界配位体的不同,铬络离子具有多种几何异构体。中心离子Cr3+,可以通过中继基的“桥合”作用,由单核变成多核,从而使络合物的分子链增长,分子变大。研究表明,具有鞣性的铬络合物必须是:(1)内界同时具有水分子、羟基、酸根等配位体(因为水分子是胶原官能团进入内界配位的取代主要对象,羟基、酸根为中继基,能“桥合”中心离子使之多核化,并能使铬络合物具有碱度或显示蒙囿作用);(2)铬络合物应为多核结构,具有足够长的分子链,能够在胶原相邻主链间形成交联键;(3)应以铬络阳离子为主,以有利于胶原的羧基进入内界与中心离子Cr3+配位。铬络合物的结构示例如下:
其他无机鞣剂几乎都能形成类似于铬的络合物结构。由于它们单独用于鞣制,成革的性能不甚理想,因而在实际生产中仅用以组合起来,进行结合鞣制或配制成异金属多核络合鞣剂(又称复合鞣剂或金属络鞣剂)使用。
几种常见的异金属双核或多核络合物主结构示例如下:
聚磷酸盐(玻璃体)的结构示例如下:
2.有机鞣剂
以植物鞣质为代表。植物鞣质化学表明,植物鞣质是一个多组分体系。根据其结构特征,曾有多元酚的衍生物之称。按化学分类法可划分为水解类鞣质(如柯子、橡椀子、栗木等)和缩合类鞣质(如荆树皮、坚木、落叶松等)。植物鞣质因类别不同,其性质各异。研究表明,植物鞣质的鞣性基团主要是酚羟基,其次是醇羟基、酚羧基、醚氧基等。鞣质酚羟基的鞣性会受到鞣质胶体微粒的分散度、电化学性质以及鞣制反应的位阻、介质条件的影响。在鞣制时,酚羟基等鞣性基团,主要作为氢键结合的吸引体和受引体与胶原相应的反应点结合。其中的醇羟基或酚羟基也可能与胶原的带电氨基形成电价键。无论是以氢键结合为主反应还是以电价键为主反应,现有的植鞣理论在承认鞣质结构中酚羟基参与鞣制反应这一点是一致的。因此,几乎都用酚羟基作为鞣质的表征。鞣质示例如下:
醛鞣剂主要包括甲醛、戊二醛、糠醛和双醛淀粉及双醛纤维素等,其中应用较广的还是甲醛和戊二醛。醛类鞣剂的结构特征是都含有醛基(—CHO)鞣性基团。醛基的结构特殊,性质活泼,反应能力很强,能与氨基化合物反应。醛鞣剂在鞣制时,其醛基参与反应,使胶原相邻的氨基通过亚甲基(—CH2—)桥,形成交联键。
油鞣剂主要包括天然动、植物油及其加工产品(如亚硫酸化鱼油)和合成油鞣剂(如烷基磺酰氯)。天然动、植物油的结构特征是含有不饱和脂肪酸上的双键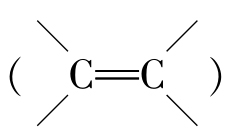 ,纯油鞣的“过氧化物”或“环氧化物”鞣制理论未被公认,更多地认为油脂是通过形成氧化物膜或氧化物聚合物沉积,包覆于胶原纤维表面的。合成油鞣剂烷基磺酰氯的鞣性基团为磺酰氯(—SO2Cl),其与胶原的碱性氨基作用,能形成砜亚胺桥(—NH—SO2—),以交联胶原相邻主链。
,纯油鞣的“过氧化物”或“环氧化物”鞣制理论未被公认,更多地认为油脂是通过形成氧化物膜或氧化物聚合物沉积,包覆于胶原纤维表面的。合成油鞣剂烷基磺酰氯的鞣性基团为磺酰氯(—SO2Cl),其与胶原的碱性氨基作用,能形成砜亚胺桥(—NH—SO2—),以交联胶原相邻主链。
合成鞣剂有芳族与脂族之分。芳族合成鞣剂主要是酚、萘类经磺化、缩合或缩合、磺化的产物,因磺化、缩合的方法和条件不同,所得产物有别。一般有辅助性和代替性之分,前者用作助剂,后者可代替部分植物鞣剂或其他鞣剂,其主要鞣性基团为酚羟基,因而鞣制性能近乎植物鞣剂。脂族合成鞣剂主要是树脂鞣剂,其常见者有氰胺树脂和脲醛树脂。前者可以三聚氰胺为例后者可以脲环树脂为例。树脂鞣剂可分为部分缩合的和完全缩合的两类,其主要鞣性基团为支链上的羟甲基,能与胶原的碱性基团形成氢键或以亚甲基桥形成共价交联键,但是更多的还是树脂本身的缩聚作用形成大分子沉积于胶原结构中,显示填充效应。
在实际生产中,除纯植物鞣、纯油鞣外,其他有机鞣剂很少单独用于鞣革,一般较多地用于结合鞣制。
化学动力学的目的是研究化学反应速度和影响反应速度的条件。通过研究鞣制化学动力学,来揭示鞣制过程的复相平衡和复相反应速度及其影响的本质,从而微观地了解鞣制反应机理,这是科学的、理想的方法。可是,由于鞣制系统的复杂性和动力学研究的难度,使得这方面的研究成为悬而未解的课题。事实上,要凭借动力学的公式进行数学演算,全面地揭示鞣制反应的历程和机理,是很难建树的。下面,仅就有关鞣制化学动力学方面的某些问题,略加阐述。
(一)关于扩散
胶体化学原理表明,胶体微粒的扩散是以布朗运动方式进行的。产生这种运动,是胶体微粒在介质中受到介质分子不均匀的撞击以及它本身的热运动的结果。布朗运动是曲折而无秩序的,但是经过一定时间,微粒还是有一个平均的位移。
平均位移Δ可以下式定量计算:
式中:D—扩散系数;t—时间。由(1)式可知,微粒平均位移的平方Δ2和位移的时间t成正比。
微粒移动的速度v,可由下式计算:
式中:F—微粒移动总力;f—介质阻力;c—微粒体积克分子浓度;N—阿伏伽德罗常数6.02×1023。由(2)式可知,微粒的移动速度v和单位微粒所需动力(即F/cN)成正比,与介质阻力f成反比。
根据菲克-爱因斯坦扩散定律,微粒的扩散量Q,可用下列公式计算:
式中:R—气体常数;N—阿伏伽德罗数;T—热力学温度;η—介质黏度;r—微粒半径;S—扩散表面;dc/dx—浓度梯度[c—浓度,x—微粒在x方向的位移(距离)]。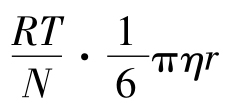 =D(扩散系数)
=D(扩散系数)
由(3)式可知,在dt时间内,微粒的扩散量Q与热力学温度T、扩散表面S、浓度梯度dc/dx成正比;同介质的黏度η、微粒半径r成反比。
若将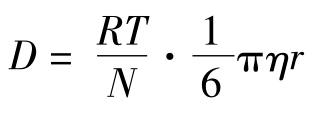 代入(3)式,则得:
代入(3)式,则得:
由(4)式可知,微粒的扩散量Q主要取决于浓度梯度。在可比的条件下(即D、S为定值),dt时间内,微粒扩散量与浓度梯度成正比。当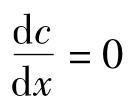 时,复相反应建立动态平衡,浓度相等,扩散作用处于相对静止。若反应保持
时,复相反应建立动态平衡,浓度相等,扩散作用处于相对静止。若反应保持 为最高值,则微粒的扩散量也达最大值。由此可见,浓度梯度是微粒扩散的动力。
为最高值,则微粒的扩散量也达最大值。由此可见,浓度梯度是微粒扩散的动力。
如上所述,微粒的扩散速度与扩散量,同扩散时间、温度、外力、浓度梯度及微粒半径等有密切关系。其中,外力、浓度梯度、微粒半径的关系尤其重要。
在实际生产中,植物鞣液是典型胶体溶液,鞣质微粒即为胶体微粒。其他鞣剂虽然单分子(或离子)质点大小属真溶液范围(d<10-7cm),但是,在配成鞣液或鞣制时,由于质点的聚集或络合物的多核化,实际上鞣剂质点都具有或近乎胶体微粒的大小(d=10-7~10-5cm)。因此,一般鞣液多为胶体多相分散体系,鞣剂质点也具有胶体微粒的某些特性。上述所讨论的微粒扩散的一般原理,也适用于鞣剂质点的扩散作用。
在鞣制过程中,鞣剂质点的布朗运动、质点的分散与聚集平衡、鞣液的浓度梯度、质点扩散的外力等,都是鞣剂质点扩散作用并借以进入皮内的重要因素和动力。实践证明,鞣制反应的速度,主要取决于鞣剂质点的扩散速度。基于此理,在无浴(少浴)快速鞣制工艺的控制上,综合运用高浓度(以保持浓度梯度)、调节pH(使质点向分散态移动)、强化机械作用(强大质点扩散外力)等技术措施。
(二)关于渗透
从渗透的定义出发,根据渗透压原理,把裸皮看作为半透膜,皮内含水,皮外为一定浓度的鞣液,那么,皮、液两相间的浓度差会使鞣剂质点向皮内渗透。这种现象本质上是一种反渗透作用。产生这种反渗透作用的条件是:裸皮的外压必须大于内压,纤维间的孔径必须大于鞣剂质点的直径。否则,渗透压的作用将导致裸皮脱水(亦即皮内的水向鞣液中渗透),鞣剂质点难以进入皮内,破坏鞣制,造成表面过鞣(或称“死鞣”)。
为了增大鞣剂质点向皮内渗透的动力,以提高鞣制速度,保持鞣液高浓度或采用干鞣工艺是有益的。因为当皮、液两相的浓度差趋近于零,则浓度将达成平衡,鞣剂质点的渗透作用近乎停止。通过鞣前预处理,使皮纤维得以分离、固定,裸皮呈多孔性,保持鞣剂质点渗透进入皮内的途径畅通,既有利加速渗透,又可防止“死鞣”。无浴快速植鞣工艺十分强调裸皮的预处理,目的在于加速渗透,防止“死鞣”。
(三)关于吸附
根据朗缪尔化学吸附原理,固态物质内部原子(或原子团)的化合价力是平衡的,但是处于表面部分的原子(或原子团),其接触空间部分存在剩余的化合价力,这种剩余价力是产生吸附作用的本质。用化学吸附原理解释鞣制反应的吸附作用,其立论的基点是把鞣剂质点作为被吸附质,而胶原作为吸附质。当鞣剂质点因扩散作用和渗透作用而进入皮内后,便分布于胶原纤维结构的固相表面,由于胶原结构表面的质点存在剩余价力,为使表里间获得平衡,就把这种剩余价力指向空间,当鞣剂质点趋近于胶原表面时,这种剩余价力就成为胶原表面对鞣剂质点的吸附力,于是鞣剂质点被吸附在胶原表面。但是被吸附的鞣剂质点会因振动而脱离胶原表面,呈现解吸作用。因而可逆的吸附过程存在着吸附与解吸的动态平衡。同时,吸附过程还要产生吸附热。这种化学吸附理论近乎表面张力效应。
化学动力学的复相反应原理,为固、液两相反应的吸附过程模拟了如下的反应机制:(1)反应分子扩散到固体表面;(2)反应分子被吸附在固体表面;(3)分子在表面上进行反应;(4)生成物从表面解吸;(5)生成物通过扩散而离开表面。
多数研究者认为,多固体与液体的反应,扩散是最慢的步骤,因此决定整个过程的速度,应为(1)(5),是否如此,尚待验证。
鞣制过程属于复相反应,胶原表面对鞣剂质点的吸附,也被引用上述吸附反应机制加以解释。但是,吸附作用并不是鞣制反应的本质,只能是鞣制反应的一个历程或非本质的一种形式。
(四)关于沉积
鞣制理论的物理学说,以沉积为鞣制效应,因而曾有“固定鞣”“沉积鞣”说。
对沉积作用的解释观点很多,最主要的是胶体微粒聚沉作用。用这种观点解释植物鞣制过程中鞣质微粒在胶原纤维表面上的沉积是合宜的。但是,在同一沉积作用问题上,围绕沉积作用的起因或发生,却众说纷纭。—说是化学吸附引起的;另说是表面张力效应的结果;有的用互溶效应加以解释,因为胶溶剂分子被胶原吸取,互溶效应被破坏,鞣剂中原来溶解的组分发生沉积;有的用胶体微粒因介质的pH变化或电解质的作用而失去表面电荷,从而变得不稳定,微粒间聚集成更大颗粒而沉降来说明。总之,承认沉积作用在鞣制反应中占有一定位置是一致的。有的研究提出,吸附、沉积等作用不一定发生于胶原与鞣剂的化学结合之前,认为鞣剂质点首先在胶原侧链方向上结合,当胶原的活性基团被饱和以后,鞣剂质点便开始吸附、沉积在胶原纤维结构内。
沉积作用同样能适用于油鞣、树脂鞣乃至于铬鞣等金属络合物的鞣制。目前对于油鞣、树脂鞣等鞣制理论,更多地重视鞣剂质点在胶原结构中的沉积作用,因为这种沉积作用在鞣制过程中所显示的填充效应已被公认,它对于成革的耐水性、丰满性和增厚作用为实践所证实。
对于沉积作用速度的定性和定量研究,目前还很少见这方面的报道。
(五)关于化学结合
鞣剂质点与胶原官能团的化学结合,是鞣制理论的精髓。化学结合以交联理论为代表。
交联理论认为,鞣制作用必须是含多官能团的鞣剂分子和胶原结构中两个或两个以上的反应点作用,形成新的分子交联键。当鞣剂分子的官能团数目和分子链长度都足以在胶原相邻分子链间形成许多交联键时,就能产生多点结合,形成形似“网络”的鞣制作用,因而显示鞣制效应。(https://www.xing528.com)
交联键的本质是化学键。鞣制作用的化学键类型包括氢键、电价键、配位键(外轨型络合物的配位键近乎电价键,内轨型络合物的配位键近乎共价键)和共价键。各种不同鞣剂与胶原反应所产生的交联键类型和数量差别很大,只有经过定性和定量的研究,才能获得可靠的结论。研究表明,以铬鞣为代表的金属络合物鞣制,主要是铬阳离子与胶原侧链上的羧基配位,所形成的交联键为配位键;以植物鞣为代表的酚系鞣剂鞣制,主要是鞣性酚羟基与胶原主链上的酰胺基(肽基)和侧链的亚氨基作用,形成的交联键多为氢键,但也可能有电价键;醛类鞣剂、磺酰氯鞣剂与胶原侧链氨基会产生缩合反应,形成的交联键为共价键。在复杂的鞣制反应中,存在分子间的作用是无疑的。
就化学键而论,电价键(即离子键)、配位键、共价键都是原子间较强的相互作用,键能为125.4~209kJ/mol,因而结合稳定而牢固。氢键严格区分起来属分子间的相互作用,与范德华力、离子的极化作用相类似,同属分子间作用力的范畴。氢键的键能较小,一般小于33.44kJ/mol,因而结合的牢固程度差。
关于鞣制反应化学结合的成键机理,可以根据化学键理论进行定性地解释。其中,电价键的形成基于离子官能团的静电作用;配位键的形成基于络合物中心离子与配位体之间的配位作用;共价键的形成基于非离子官能团之间成键电子的电子云重叠。氢键的形成基于分子间的极性作用。
鞣制反应的交联成键,不仅同胶原的官能团反应活性有关,而且与鞣剂分子的结构(如分子大小、空间构型、位阻作用等)、性质(如碱度、收敛性、电荷性质等)密切相关,同时介质条件也会产生影响。因此,企图用动力学研究复杂的、瞬时的鞣制反应成键过程和速度,无疑是十分困难的。
根据鞣制化学动力学的研究,鞣制反应的历程可模拟成如下的假设步骤和模式:
(1)鞣剂质点以扩散作用进入裸皮内部;(2)鞣剂质点以渗透作用进入裸皮内部;(3)鞣剂质点被吸附在胶原纤维表面;(4)鞣剂质点沉积在胶原纤维表面;(5)被吸附、沉积的鞣剂质点与胶原发生化学结合。
假设步骤间的联系模式为:
基于上述,为了便于理解,对鞣制反应的历程,可作如下阐述:
在鞣制过程中,作为反应主体的鞣剂与胶原间的互相作用,主要表现为渗透与结合两个方面,这是鞣制全过程中的一对主要矛盾。它们的存在、发展和转化,决定鞣制反应的历程。“一切矛盾都依一定的条件向它们的反面转化着”(《矛盾论》)。鞣制初期,鞣剂质点与裸皮刚开始接触,鞣剂质点以布朗运动、外力和裸皮内、外鞣液的浓度差为动力,向皮内扩散、渗透,此时渗透处于主导地位,结合从属之。随着鞣剂质点相继渗透,大量鞣剂质点进入裸皮深处,分布于胶原纤维结构内,并陆续被胶原纤维表面所吸附(吸附与解吸存在可逆平衡)或因聚沉作用沉积在纤维间。于是,鞣液中鞣剂质点减少,浓度降低,皮、液两相的浓度渐小,直至浓度相等,鞣制反应建立平衡,则渗透与结合并存,彼此处于相持地位,动力学上称为对峙反应,此时应为鞣制中期。“一切平衡都只是相对的和暂时的”(《自然辩证法》)。随着鞣制条件的改变(如铬鞣提碱、植鞣提高鞣液浓度等)和大分子多官能团的鞣剂质点与胶原相邻结构之间单点或多点结合的产生,结合开始上升为主导地位。由于皮内剩余的自由鞣剂质点因结合而减少,旧的浓度平衡被破坏,促使鞣液中所剩的鞣剂质点仍向皮内渗透,以建立新平衡。尽管渗透仍在持续,但与结合相比,渗透的量和速度甚微。因为鞣制进入后期,渗透已转化为从属地位。总之,在鞣制过程中,渗透与结合是互为依存的,渗透过程中伴随着结合,结合过程中持续着渗透。当鞣剂质点渗透加大,其与胶原的结合就会自然地增加;由于结合量的逐步提高,自由扩散的剩余鞣剂质点渐少,渗透速度也就相应降低。但是,渗透以结合为目的,结合则以渗透为前提。
鞣剂质点与胶原实现结合,意味着鞣制反应的完成。就结合而言,鞣剂质点与胶原结合是鞣制反应的共性;不同类型的鞣剂与胶原实现不同形式的结合,则是鞣制反应的个性。共性寓于个性之中。所以分别讨论各种鞣制反应的结合形式是有意义的。
(一)铬鞣
铬鞣机理基于络合物的配位理论,铬鞣反应取决于铬络合物的组成和性质。只有当胶原相邻分子链上的官能团(主要是羧基,也包括不带电的氨基)进入铬络合物内界,取代不稳定的配位体(如水分子),与中心离子Cr3+以配位键形成双点结合(同时存在大量单点结合),“桥合”了胶原结构,才能显示铬鞣效应。
根据X-衍射照片研究和电子显微镜下观察,从铬鞣过程胶原的原纤维变化中,证实铬络合物与胶原分子在侧链上发生定向结合,并不涉及主链上的肽键。
有的研究指出胶原的谷氨酸、门冬氨酸的残基参与同铬盐的结合,这就证实了羧基配位的确切性。
离子交换法和测定皮革中结合的铬及酸根的计算结果表明,当碱度为33%的硫酸铬络合物与胶原作用达到平衡后,Cr2O3的结合量为11%,其中单点结合占90%,双点结合占10%。
(1)多核铬络阳离子与胶原相邻羧基分别成六节环配位的双点结合见反应式Ⅰ。
(2)双核铬络阳离子与胶原相邻羧基以配位键、氢键的双点结合见反应式Ⅱ。
注:胶原相邻羧基分别与两个Cr3+配位,同时羧基上的羰基与配位羟基形成氢键。
(3)双核铬络阳离子与胶原相邻官能团以配位键、氢键的多点结合见反应式Ⅲ。
注:①为相邻羧基阴离子与两个Cr3+配位;
②为相邻不带电氨基与两个Cr3+配位;
③为相邻羧基上的羰基与配位水分子的氢形成氢键。
(4)双核铬络阴离子与胶原相邻氨基阳离子分别以电价键的双点结合见反应式Ⅳ。
说明:①Ⅰ式为铬鞣的主反应,为了简便,Ⅱ、Ⅲ、Ⅳ式以双核铬络离子表示,实际反应更接近Ⅰ式;Ⅳ式为阴离子蒙囿剂强蒙囿铬鞣;
②单点结合可理解为上述双点结合之一侧;
③Ⅰ式络离子内界未表示出硫酸根,特意保留这种特殊性。
研究和实践均已证明,铬鞣具有无机鞣剂(金属络合物等)鞣制的代表性,因此探讨铬鞣结合形式,对于了解铝鞣、铁鞣、锆鞣、钛鞣以及异金属多核络合鞣剂鞣制的反应和结合形式,具有典型意义。所以,不一一赘述。
(二)植物鞣
植物鞣机理更多地倾向于氢键作用理论,许多研究者主张以鞣质的酚羟基与胶原主链上的酰胺基(肽基)所形成的氢键结合为主反应。无论是水解类鞣质还是缩合类鞣质,其参与植鞣反应的主要鞣性基团是酚羟基。植鞣反应取决于鞣质的结构及其胶体微粒的分散度、电化学等性质。在肯定植鞣反应以鞣质酚羟基与胶原主链上的—NH—CO—键以氢键结合为主的同时,认为胶原侧链氨基上形成电价键、共价键也是可能的,对于鞣质微粒在胶原结构表面的沉积作用和填充效应,也得到应有重视。
电子显微镜的观察和X-射线结构分析表明,鞣质的结合既在肢原肽键位置上发生,又在胶原侧链方向上发生,同时,还发现约有1/5的肽键被鞣质微粒所占据。
研究者通过聚酰胺纤维为胶原主链的模拟物和封闭胶原侧链的碱性氨基后再行植鞣实验,验证了主链上的氢键结合与侧链上的电价键结合的确切性。有的研究指出,鞣质与胶原碱性基的结合位置,主要在胶原结构中赖氨酸、精氨酸的残基上。其中,水解类鞣质以羧基(—COOH)与之形成电价键,而缩合类鞣质则可能以酚羟基(—OH)与之形成共价键。
植鞣反应式Ⅴ如下:
(三)结合鞣
结合鞣的意义在于利用各种鞣剂的特殊性能,取长补短,以达到各种要求。由于胶原是多官能化合物,侧链上的氨基酸残基的反应活性是多方面的。为了适应某种需要,可选用具有多种反应性的不同鞣剂,同时或先后参与鞣制,使胶原相应的位置上获得综合鞣制的效果,以提高和改善成革的性能。近年来,为了节约代用红矾、减少污染和公害,改革工艺,提高成革质量,结合鞣得到普遍重视,实验和投产的工艺方案大批涌现,就其形式主要包括预鞣、主鞣、复鞣、结合鞣(又称混合鞣),工艺方案因选用的鞣剂及生产品种不同而异。
(1)铬-植结合鞣主反应见反应式Ⅵ。
(2)铝-铬结合鞣主反应见反应式Ⅶ。
(3)铝-铬-铁结合鞣主反应见反应式Ⅷ。
(4)脲环-铝铬结合鞣主反应见反应式Ⅸ。
(5)钛-锆结合鞣主反应见反应式Ⅹ。
(6)醛-铝结合鞣主反应见反应式Ⅺ。
(7)油-铝-铬结合鞣主反应见反应式Ⅻ。
注:(1)铬-植结合鞣机理近乎纯铬鞣与纯植鞣,Ⅵ式仅简略示意,植物鞣质与胶原侧链氨基成电价结合,不一定是主反应。
(2)铝-铬结合鞣所用的铝-铬络合鞣剂的配比Cr2O3∶Al2O3=(4~5)∶1,Ⅶ式仅以Cr∶Al=1:1的双核络合物示意。
(3)铝-铬-铁结合鞣一般先铝预鞣,再以铬-铁络合鞣剂主鞣,鞣制反应有可借中继基(OH,SO4)桥合成多核络合物,Ⅷ式仅是可能的示意式,FeSO4·7H2O∶Na2Cr2O7·2H2O=(6~8)∶(1.0~1.3)。
(4)Ⅸ式中脲环上的羟甲基与胶原侧链氨基形成氢键,实际上主反应也可能发生在胶原相邻主链的肽基位置上;其他氨基树脂(如三聚氰胺预聚体)亦然;
(5)钛-锆结合鞣据成都工学院皮革系资料介绍,鞣剂的比例为:TiO2∶ZrO2=2:1;该资料介绍,纯钛鞣机理研究的结论是:在钛鞣剂溶液中主要是阴离子络合物。研究表明,钛鞣剂主要和胶原模型(聚酰胺纤维、离子交换剂)中的羟基、碱性基和羧基反应,还可能和醇羟基、肽基反应。Ⅹ式是基于上述研究并结合锆鞣剂的结构与性质所推测的反应示意式;
(6)资料介绍,甲醛与胶原含氮基反应强弱次序为:ε-氨基>α-氨基>胍基;甲醛能自身聚合,无论是胶原侧链还是主链之间,其聚合体的分子链长度足以满足空间距离的要求,见Ⅺ式。
(7)油-铝-铬结合鞣中,油鞣剂组分多为加脂剂和磺酰氯,加脂剂近乎纯油鞣,主要表现为沉积作用,而磺酰氯能与胶原碱性基成共价键结合,Ⅻ式为烷基双磺酰氯与胶原侧链氨基交联,其中R约为C18。
学习理论的目的全在于应用。探讨鞣制机理,目的在于从鞣制的宏观现象中获得微观认识,理解鞣制过程的物理化学本质,从而掌握制定鞣制工艺方案和控制鞣制过程的主动权。实践和理论同样表明,采用无浴(少浴)速鞣工艺,关键在于提高鞣剂的渗透速度和增加鞣剂质点与胶原的结合量。结合鞣法在速鞣工艺中的潜在力量,也应当引起重视。
(原载《皮革科技》1980,第1期、第2期)
[1]上海市轻工业学校编.制革工艺学[M].北京:轻工业出版社,1965.
[2]《无机化学》编写组编.无机化学[M].北京:人民教育出版社,1978.
[3]黄子卿著.物理化学[M].人民教育出版社,1955.
[4]河南皮革研究所.国外制革工业水平[J].皮革科技动态,1977,10.
[5]魏庆元.国外铬盐鞣革的研究资料述评[J].皮革科技动态,1976,9.
[6]张文德,蒋廷方.植物鞣质的鞣革理论[J].四川皮革,1978,2.
[7]徐士弘,蒋廷方.钛盐鞣革性能文献综述.成都工学院《皮革科技》参考资料汇编(三)
[8]马燮芳,俞志洪.酶法脱毛生产鞋正面革应用鞣制工艺的文献资料综述[J].皮革科技动态,1975:7-9.
[9]王树声,沈征复,编译.超显微技术用于皮革研究[J].皮革科技动态,1977:7-8.
[10]吕绪庸.提高底革耐磨性的技术理论探讨[J].皮革科技动态,1977.8.
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。